General Information About Ewing Sarcoma and Undifferentiated Small Round Cell Sarcomas of Bone and Soft Tissue
Dramatic improvements in survival have been achieved for children and adolescents with cancer.[1] Between 1975 and 2020, childhood cancer mortality decreased by more than 50%.[1,2,3] For Ewing sarcoma, the 5-year survival rate has increased from 59% to a range of 80% to 85% for children younger than 15 years and from 20% to 69% for adolescents aged 15 to 19 years.[1,2]
Studies using immunohistochemical markers,[4] cytogenetics,[5,6] molecular genetics, and tissue culture [7] indicate that Ewing sarcoma originates from a primordial bone marrow–derived mesenchymal stem cell.[8,9] Older terms such as peripheral primitive neuroectodermal tumor, Askin tumor (Ewing sarcoma of chest wall), and extraosseous Ewing sarcoma (often combined in the term Ewing sarcoma family of tumors) refer to this same tumor.
The World Health Organization (WHO) classification of tumors of soft tissue and bone was modified in 2020 to introduce a new chapter on undifferentiated small round cell sarcomas of bone and soft tissue. This WHO chapter consists of Ewing sarcoma and three main categories, including round cell sarcomas with EWSR1::non-ETS fusions, CIC-rearranged sarcoma, and sarcomas with BCOR genetic alterations.[10]
Before the widespread availability of genomic testing, Ewing sarcoma was identified by the appearance of small, round, blue cells on light microscopic examination, along with positive staining for CD99 by immunohistochemistry. The identification of the recurring t(11;22) translocation in most Ewing sarcoma tumors led to the discovery that most tumors classified as Ewing sarcoma had a translocation that juxtaposed a portion of the EWSR1 gene to a portion of a gene in the ETS family, resulting in a transforming transcript. Not all undifferentiated small round cell sarcomas of bone and soft tissue have such a translocation. Further research identified additional genetic changes, including tumors with translocations of the CIC gene or the BCOR gene. These groups of tumors occur much less frequently than Ewing sarcoma, and data on these patients are based on smaller sample sizes and less homogeneous treatment; therefore, patient outcomes are harder to quantify with precision. Most of these tumors have been treated with regimens designed for Ewing sarcoma, and the consensus was that they were often included in clinical trials for the treatment of Ewing sarcoma, sometimes referred to as translocation-negative Ewing sarcoma. It is now agreed that these tumors are sufficiently different from Ewing sarcoma and that they should be stratified and analyzed separately from Ewing sarcoma, even if they are treated with similar therapy. In this summary, these tumors are described separately. For more information about these smaller groups of tumors, see the following sections:
Incidence
In the United States between 2016 and 2020, the National Childhood Cancer Registry (NCCR) reported an incidence rate of Ewing sarcoma and related sarcomas of bone of 3.0 cases per 1 million in children and adolescents younger than 20 years.[2] This incidence is unchanged from that reported between 1973 and 2004.[11] The incidence rates by age groups in the U.S. pediatric population for Ewing sarcoma and related sarcomas of bone are shown in Table 1 and Figure 1. While well-characterized cases of Ewing sarcoma in neonates and infants have been described, the incidence is low in infants and young children and then increases in adolescents.[12,13]
Table 1. 5-Year Age-Adjusted Incidence Rates for Ewing Sarcoma by Age (2016–2020)a| Age (years) | Rate per 1,000,000 | 95% Confidence Interval |
|---|
| a Source: National Childhood Cancer Registry (NCCR) Explorer.[2] |
| <1 | 0.5 | 0.2–1.1 |
| 1–4 | 1 | 0.7–1.3 |
| 5–9 | 2.3 | 1.9–2.6 |
| 10–14 | 4.3 | 3.9–4.9 |
| 15–19 | 4.5 | 4.0–5.0 |
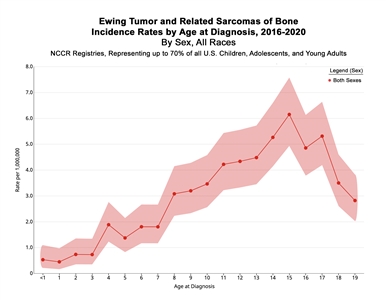
Figure 1. Incidence rates of Ewing tumor and related sarcomas of bone by age at diagnosis in the National Childhood Cancer Registry (NCCR) from 2016 to 2020. Credit: NCCR*Explorer: An interactive website for NCCR cancer statistics [Internet]. National Cancer Institute; 2023 Sep 7. [updated: 2023 Sep 8; cited 2024 Sep 4]. Available from: https://nccrexplorer.ccdi.cancer.gov.
The incidence of Ewing sarcoma in the United States is nine times greater in White people than in Black people, with an intermediate incidence in Asian people.[14,15] The relative paucity of Ewing sarcoma in people of African or Asian descent may be explained, in part, by a specific polymorphism in the EGR2 gene.[16]
Based on data from 1,426 patients entered on European Intergroup Cooperative Ewing Sarcoma Studies, 59% of patients are male and 41% are female.[17] These results match the 58%-to-42% male-to-female distribution in the United States (age <20 years) in the NCCR dataset (3.5 and 2.5 cases per million incidence rate for males and females, respectively).[2]
Genetic Predisposition to Ewing Sarcoma
Conventional understanding of translocation-driven sarcoma such as Ewing sarcoma suggests that these patients do not have a genetic predisposition.[18] A retrospective European-focused and panancestry case-controlled analysis was performed. The purpose of this study was to screen for enrichment of pathogenic germline variants in 141 known cancer predisposition genes in 1,147 pediatric patients diagnosed with sarcomas (226 Ewing sarcomas, 438 osteosarcomas, 180 rhabdomyosarcomas, and 303 other sarcomas), and compared the results to identically processed cancer-free control individuals. A distinct pattern of pathogenic germline variants was seen in Ewing sarcoma compared with other sarcoma types. FANCC was the only gene with an enrichment signal for heterozygous pathogenic variants in the European Ewing sarcoma discovery cohort (three individuals; odds ratio [OR], 12.6; 95% confidence interval [CI], 3.0–43.2; P = .003; false discovery rate, 0.40). This enrichment in FANCC heterozygous pathogenic variants was again observed in the European Ewing sarcoma validation cohort (three individuals; OR, 7.0; 95% CI, 1.7–23.6; P = .014).
Genome-wide association studies have identified susceptibility loci for Ewing sarcoma at 1p36.22, 10q21, and 15q15.[16,19,20] Deep sequencing through the 10q21.3 region identified a polymorphism in the EGR2 gene, which appears to cooperate with and magnify the enhanced activity of the gene product of the EWSR1::FLI1 fusion gene that is seen in most patients with Ewing sarcoma.[16] The polymorphism associated with the increased risk is found at a much higher frequency in White people than in Black or Asian people, possibly contributing to the epidemiology of the relative infrequency of Ewing sarcoma in the latter populations. Three new susceptibility loci have been identified at 6p25.1, 20p11.22, and 20p11.23.[20]
Clinical Presentation
Clinical presentation of Ewing sarcoma varies and depends on the tumor's size and location.
Primary sites of bone disease are listed in Table 2.[21]
Table 2. Incidence Rates of Primary Sites of Bone Disease| Primary Site | Incidence Rate |
|---|
| Skull | 5% |
| Spine | 7% |
| Rib | 11% |
| Sternum, scapula, and clavicle | 5% |
| Humerus | 7% |
| Radius, ulna, hand | 2% |
| Pelvis | 18% |
| Femur | 11% |
| Tibia, fibula, patella, foot | 14% |
| Soft tissue | 19% |
The time from the first symptom to diagnosis of Ewing sarcoma is often long, with a median interval reported from 2 to 5 months. Longer times are associated with older age and pelvic primary sites. Time from the first symptom to diagnosis has not been associated with metastasis, surgical outcome, or survival.[22]
Approximately 25% of patients with Ewing sarcoma have metastatic disease at the time of diagnosis, with lung, bone, and bone marrow being the most common metastatic sites.[11]
A retrospective analysis examined patients treated on two Children's Oncology Group (COG) studies, INT-0154 and AEWS0031 (NCT00006734). This study compared the clinical characteristics of 213 patients with extraskeletal primary Ewing sarcoma with those of 826 patients with primary Ewing sarcoma of bone.[23] Patients with extraskeletal tumors were more likely to be non-White, have axial primary tumors, and have smaller tumors than patients with primary Ewing sarcoma of bone.
The Surveillance, Epidemiology, and End Results (SEER) Program database was used to compare patients younger than 40 years with Ewing sarcoma who presented with skeletal and extraosseous primary sites (see Table 3).[24] Patients with extraosseous Ewing sarcoma were more likely to be older, female, of non-White race, and have axial primary sites, and they were less likely to have pelvic primary sites than were patients with skeletal Ewing sarcoma.
Table 3. Characteristics of Patients With Extraosseous Ewing Sarcoma and Skeletal Ewing Sarcomaa| Characteristic | Extraosseous Ewing Sarcoma | Skeletal Ewing Sarcoma | P Value |
|---|
| a Adapted from Applebaum et al.[24] |
| Mean age (range), years | 20 (0–39) | 16 (0–39) | <.001 |
| Male | 53% | 63% | <.001 |
| White race | 85% | 93% | <.001 |
| Axial primary sites | 73% | 54% | <.001 |
| Pelvic primary sites | 20% | 27% | .001 |
Diagnostic Evaluation
The following tests and procedures may be used to diagnose or stage Ewing sarcoma:
- Physical examination and history.
- Magnetic resonance imaging (MRI) of primary tumor site.
- Computed tomography (CT) scan of chest.
- Positron emission tomography (PET) scan.
- Bone scan. Bone scan was traditionally routinely performed on all patients with Ewing sarcoma for staging. However, many investigators believe that the PET scan can replace the bone scan.[25,26]
- Bone marrow aspiration and biopsy.
- X-ray of primary bone sites.
- Complete blood count.
- Blood chemistry studies, such as lactate dehydrogenase (LDH).
Skip metastasis evaluation is important for primary appendicular bone tumors. Thus, imaging of the entire involved bone is standardly performed. In one retrospective study, skip metastasis was seen in 15.8% of patients. The presence of skip metastasis was associated with an increased risk of distant metastatic disease.[27]
Omission of bone marrow biopsy and aspiration may be considered, when fluorine F 18-fludeoxyglucose (18F-FDG) PET imaging is used, in patients with otherwise localized disease after initial staging studies. A systematic review of Ewing sarcoma studies was performed to assess the incidence of bone marrow metastasis and the role of 18F-FDG PET imaging to detect bone marrow metastasis.[28] The review reported a pooled incidence of bone marrow metastasis of 4.8% in all patients with newly diagnosed Ewing sarcoma and 17.5% in patients with metastatic disease. Only 1.2% of patients had bone marrow metastasis as their sole metastatic site. Compared with bone marrow biopsy and aspiration, 18F-FDG PET detection of bone marrow metastasis demonstrated pooled 100% sensitivity and 96% specificity, positive predictive value of 75%, and negative predictive value of 100%. For more information about diagnostic biopsy, see the Treatment Option Overview for Ewing Sarcoma section.
Prognostic Factors
The two major types of prognostic factors for patients with Ewing sarcoma are grouped as follows:
Pretreatment factors
- Metastases: The presence or absence of metastatic disease is the single most powerful predictor of outcome. Any metastatic disease defined by standard imaging techniques or bone marrow aspirate/biopsy by morphology is an adverse prognostic factor. Metastases at diagnosis are detected in about 25% of patients.[11]
Patients with metastatic disease confined to the lung have a better prognosis than patients with extrapulmonary metastatic sites.[29,30,31,32] The number of pulmonary lesions does not seem to correlate with outcome, but patients with unilateral lung involvement have a better prognosis than patients with bilateral lung involvement.[33]
Patients with metastasis to only bone seem to have a better outcome than patients with metastases to both bone and lung.[34,35]
Based on an analysis from the SEER database, regional lymph node involvement in patients is associated with an inferior overall outcome when compared with patients without regional lymph node involvement.[36]
- Site of tumor: Patients with Ewing sarcoma in the distal extremities have more favorable outcomes. Patients with Ewing sarcoma in the proximal extremities have an intermediate prognosis, followed by patients with central or pelvic sites.[29,31,32,37] However, a trial from the COG showed similar outcomes for patients with pelvic primary tumors compared with other sites.[21]
One study retrospectively analyzed a single-institution's experience with visceral Ewing sarcoma. The study focused on surgical management and compared the outcomes of patients with visceral Ewing sarcoma with those of patients with osseous and soft tissue Ewing sarcoma.[38] There were 156 patients with Ewing sarcoma identified: 117 osseous Ewing sarcomas, 20 soft tissue Ewing sarcomas, and 19 visceral Ewing sarcomas. Visceral Ewing sarcomas arose in the kidneys (n = 5), lungs (n = 5), intestines (n = 2), esophagus (n = 1), liver (n = 1), pancreas (n = 1), adrenal gland (n = 1), vagina (n = 1), brain (n = 1), and spinal cord (n = 1). Visceral Ewing sarcoma was more frequently metastatic at presentation (63.2%; P = .005). However, there was no significant difference in overall survival (OS) or relapse-free survival among the Ewing sarcoma groups, with similar follow-up intervals.
- Extraskeletal versus skeletal primary tumors: The COG performed a retrospective analysis from two large cooperative trials that used similar treatment regimens.[23] They identified 213 patients with extraskeletal primary tumors and 826 patients with skeletal primary tumors. Patients with extraskeletal primary tumors were more likely to have an axial primary site, less likely to have large primary tumors, and had a statistically significant better prognosis than did patients with skeletal primary tumors.
- Tumor size or volume: Most studies have shown that tumor size or volume is an important prognostic factor. Cutoffs of a volume of 100 mL or 200 mL and/or single dimension greater than 8 cm are used to define larger tumors. Larger tumors tend to occur in unfavorable sites.[31,32,39]
- Age: Younger patients generally have a better prognosis than older patients, as noted in the following studies:[13,29,32,37,40,41,42]
- In North American studies, patients younger than 10 years had a better outcome than those aged 10 to 17 years at diagnosis (relative risk [RR], 1.4). Patients older than 18 years had an inferior outcome (RR, 2.5).[43,44,45]
- A retrospective review of two consecutive German trials for Ewing sarcoma identified 47 patients older than 40 years.[46] With adequate multimodal therapy, survival was comparable to the survival observed in adolescents treated on the same trials.
- Review of the SEER database from 1973 to 2011 identified 1,957 patients with Ewing sarcoma.[47] Thirty-nine of these patients (2.0%) were younger than 12 months at diagnosis. Infants were less likely to receive radiation therapy and more likely to have soft tissue primary sites. Early death was more common in infants, but the OS did not differ significantly from that of older patients.
- A European retrospective review identified 2,635 patients with Ewing sarcoma of bone.[48] Sites of primary and metastatic tumors differed according to the age groups of young children (0–9 years), early adolescence (10–14 years), late adolescence (15–19 years), young adults (20–24 years), and adults (older than 24 years). Young children had the most striking differences in site of disease, with a lower proportion of pelvic primary and axial tumors. Young children also presented less often with metastatic disease at diagnosis.
- Sex: Females with Ewing sarcoma have a better prognosis than males with Ewing sarcoma.[14,32,37]
- Serum LDH: Increased serum LDH levels before treatment are associated with inferior prognosis. Increased LDH levels are also associated with large primary tumors and metastatic disease.[37]
- Pathological fracture: A single-institution retrospective analysis of 78 patients with Ewing sarcoma suggested that pathological fracture at initial presentation was associated with inferior event-free survival (EFS) and OS.[49][Level of evidence C1] Another study found that pathological fracture at the time of diagnosis did not preclude surgical resection and was not associated with an adverse outcome.[50]
- Previous treatment for cancer: In the SEER database, 58 patients with Ewing sarcoma were diagnosed after treatment for a previous malignancy (2.1% of patients with Ewing sarcoma). These patients were compared with 2,756 patients with Ewing sarcoma as a first cancer over the same period. Patients with Ewing sarcoma as a second malignant neoplasm were older (secondary Ewing sarcoma, mean age of 47.8 years; primary Ewing sarcoma, mean age of 22.5 years), more likely to have a primary tumor in an axial or extraskeletal site, and had a worse prognosis (5-year OS rates of 43.5% for patients with secondary Ewing sarcoma and 64.2% for patients with primary Ewing sarcoma).[51]
- Chromosomal alterations:
- Complex karyotype (defined as the presence of five or more independent chromosome abnormalities at diagnosis) and modal chromosome numbers lower than 50 appear to have adverse prognostic significance.[52]
- Gain of chromosome 1q and/or deletion of chromosome 16q has been associated with inferior prognosis for patients with Ewing sarcoma in several cohorts.[53,54,55] These two chromosomal alterations commonly occur together across a range of cancer types, including Ewing sarcoma.[56] Their co-occurrence is likely a result of their derivation from an unbalanced t(1;16) translocation resulting in gain of chromosome 1q together with loss of chromosomal material from 16q.[57,58]
- Detectable Ewing sarcoma cells, fusion transcripts, or circulating tumor DNA (ctDNA) in peripheral blood: Several techniques to evaluate the presence of Ewing sarcoma in the peripheral blood have been proposed. Flow cytometry for cells that express the CD99 antigen was not sufficiently sensitive to serve as a reliable biomarker.[59,60] Reverse transcriptase–polymerase chain reaction (RT-PCR) for the EWSR1::FLI1 translocation was also not considered a reliable biomarker.[61]
A more sensitive technique used patient-specific primers designed after identification of the specific translocation breakpoint in combination with droplet digital PCR to detect the EWSR1 fusion. This technique reported a sensitivity threshold of 0.009% to 0.018%.[62] Levels of circulating cell-free DNA were higher in patients with metastatic disease than in patients with localized disease.
A next-generation sequencing hybrid capture assay and an ultra-low-pass whole-genome sequencing assay were used to detect the EWSR1 fusion in ctDNA in banked plasma from patients with Ewing sarcoma. Among patients with newly diagnosed localized Ewing sarcoma, detectable ctDNA was associated with inferior 3-year EFS rates (48.6% vs. 82.1%; P = .006) and OS rates (79.8% vs. 92.6%; P = .01).[63]
ctDNA was separately assayed by digital-droplet PCR in 102 patients who were treated in the EWING2008 (NCT00987636) trial.[64] Pretreatment ctDNA copy numbers correlated with EFS and OS. A reduction in ctDNA levels below the detection limit was observed in most patients after only two blocks of vincristine, ifosfamide, doxorubicin, and etoposide (VIDE) induction chemotherapy. The persistence of ctDNA after two blocks of VIDE was a strong predictor of poor outcomes.
- Detectable fusion transcripts in morphologically normal marrow: RT-PCR can be used to detect fusion transcripts in bone marrow. In a single retrospective study using patients with normal marrow morphology and no other metastatic site, fusion transcript detection in marrow or peripheral blood was associated with an increased risk of relapse.[60] However, a larger cohort (n = 225) of patients with localized Ewing sarcoma did not show a difference in EFS or OS based on the detection of fusion transcripts in blood or bone marrow.[65]
- Gene alterations: A prospective analysis of TP53 variants and/or CDKN2A deletions was done in patients with Ewing sarcoma enrolled on COG clinical trials. The analysis found no association of these alterations with EFS.[66]
In a study of 299 patients with Ewing sarcoma, 41 patients (14%) had STAG2 variants and 16 patients (5%) had TP53 variants.[55] There was no association with OS for patients with either the STAG2 or TP53 variant alone. However, the nine patients (3%) with tumors that had both STAG2 and TP53 variants had a significantly decreased OS rate (<20% at 4 years).
The COG analyzed STAG2 expression by immunohistochemistry in children with Ewing sarcoma who participated in frontline treatment trials.[67] STAG2 was lost in 29 of 108 patients with localized disease and in 6 of 27 patients with metastatic disease. Among patients who had immunohistochemistry and sequencing performed, no cases (0 of 17) with STAG2 expression had STAG2 variants, and 2 of 7 cases with STAG2 loss had STAG2 variants. Among patients with localized disease, the 5-year EFS rate was 54% (95% CI, 34%–70%) for those with STAG2 loss, compared with 75% (95% CI, 63%–84%) for those with STAG2 expression (P = .0034).
The following are not considered to be adverse prognostic factors for Ewing sarcoma:
- Histopathology: The degree of neural differentiation is not a prognostic factor in Ewing sarcoma.[68,69]
- Fusion subtype: The EWSR1::ETS translocation associated with Ewing sarcoma can occur at several potential breakpoints in each of the genes that join to form the novel segment of DNA. Once thought to be significant,[70] two large series have shown that the EWSR1::ETS translocation breakpoint site is not an adverse prognostic factor.[71,72]
Response to initial therapy factors
Multiple studies have shown that patients with minimal or no residual viable tumor after presurgical chemotherapy have a significantly better EFS than do patients with larger amounts of viable tumor.[21,73,74,75,76]; [77][Level of evidence C2] In particular, patients with localized disease who have no viable tumor seen at the time of local-control surgery appear to have markedly favorable outcomes.[21]; [77][Level of evidence C2] Female sex and younger age predict a good histological response to preoperative therapy.[78] For patients who receive preinduction- and postinduction-chemotherapy PET scans, decreased PET uptake after chemotherapy correlated with good histological response and better outcome.[79,80,81]
Patients with poor response to presurgical chemotherapy have an increased risk of local recurrence.[82]
A retrospective analysis of risk factors for recurrence was performed in patients who received initial chemotherapy and underwent surgical resection of the primary tumor.[83][Level of evidence C1] Among 982 patients with a median follow-up of 7.6 years, the following was reported:
- Adverse risk factors for local recurrence were pelvic primary tumors (hazard ratio [HR], 2.04; 95% CI, 1.10–3.80) and marginal/intralesional resection (HR, 2.28; 95% CI, 1.25–4.16). The addition of radiation therapy was associated with improved outcome (HR, 0.52; 95% CI, 0.28–0.95).
- Adverse risk factors for developing new pulmonary metastasis were less than 90% necrosis (HR, 2.13; 95% CI, 1.13–4.00) and previous pulmonary metastasis (HR, 4.90; 95% CI, 2.28–8.52).
- Adverse risk factors for death included pulmonary metastasis (HR, 8.08; 95% CI, 4.01–16.29), bone or other metastasis (HR, 10.23; 95% CI, 4.90–21.36), and less than 90% necrosis (HR, 6.35; 95% CI, 3.18–12.69).
- Early local recurrence (0–24 months) negatively influenced survival (HR, 3.79; 95% CI, 1.34–10.76).
In a retrospective cohort of 148 patients with pulmonary metastatic Ewing sarcoma, 41.2% had radiographic resolution of lung nodules after initial induction chemotherapy.[84] These patients had superior OS compared with patients who had residual nodules at end-induction (71.2% vs. 50.2% at 5 years). Particularly favorable outcomes were seen in the patients who had early clearance of lung nodules and received consolidative whole-lung radiation therapy (5-year OS rate, 85.2%).
References:
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
- National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed August 23, 2024.
- Surveillance Research Program, National Cancer Institute: SEER*Explorer: An interactive website for SEER cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed September 5, 2024.
- Olsen SH, Thomas DG, Lucas DR: Cluster analysis of immunohistochemical profiles in synovial sarcoma, malignant peripheral nerve sheath tumor, and Ewing sarcoma. Mod Pathol 19 (5): 659-68, 2006.
- Delattre O, Zucman J, Melot T, et al.: The Ewing family of tumors--a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 331 (5): 294-9, 1994.
- Dagher R, Pham TA, Sorbara L, et al.: Molecular confirmation of Ewing sarcoma. J Pediatr Hematol Oncol 23 (4): 221-4, 2001.
- Llombart-Bosch A, Carda C, Peydro-Olaya A, et al.: Soft tissue Ewing's sarcoma. Characterization in established cultures and xenografts with evidence of a neuroectodermic phenotype. Cancer 66 (12): 2589-601, 1990.
- Suvà ML, Riggi N, Stehle JC, et al.: Identification of cancer stem cells in Ewing's sarcoma. Cancer Res 69 (5): 1776-81, 2009.
- Tirode F, Laud-Duval K, Prieur A, et al.: Mesenchymal stem cell features of Ewing tumors. Cancer Cell 11 (5): 421-9, 2007.
- Choi JH, Ro JY: The 2020 WHO Classification of Tumors of Soft Tissue: Selected Changes and New Entities. Adv Anat Pathol 28 (1): 44-58, 2021.
- Esiashvili N, Goodman M, Marcus RB: Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. J Pediatr Hematol Oncol 30 (6): 425-30, 2008.
- Kim SY, Tsokos M, Helman LJ: Dilemmas associated with congenital ewing sarcoma family tumors. J Pediatr Hematol Oncol 30 (1): 4-7, 2008.
- van den Berg H, Dirksen U, Ranft A, et al.: Ewing tumors in infants. Pediatr Blood Cancer 50 (4): 761-4, 2008.
- Jawad MU, Cheung MC, Min ES, et al.: Ewing sarcoma demonstrates racial disparities in incidence-related and sex-related differences in outcome: an analysis of 1631 cases from the SEER database, 1973-2005. Cancer 115 (15): 3526-36, 2009.
- Beck R, Monument MJ, Watkins WS, et al.: EWS/FLI-responsive GGAA microsatellites exhibit polymorphic differences between European and African populations. Cancer Genet 205 (6): 304-12, 2012.
- Grünewald TG, Bernard V, Gilardi-Hebenstreit P, et al.: Chimeric EWSR1-FLI1 regulates the Ewing sarcoma susceptibility gene EGR2 via a GGAA microsatellite. Nat Genet 47 (9): 1073-8, 2015.
- Paulussen M, Craft AW, Lewis I, et al.: Results of the EICESS-92 Study: two randomized trials of Ewing's sarcoma treatment--cyclophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in high-risk patients. J Clin Oncol 26 (27): 4385-93, 2008.
- Gillani R, Camp SY, Han S, et al.: Germline predisposition to pediatric Ewing sarcoma is characterized by inherited pathogenic variants in DNA damage repair genes. Am J Hum Genet 109 (6): 1026-1037, 2022.
- Postel-Vinay S, Véron AS, Tirode F, et al.: Common variants near TARDBP and EGR2 are associated with susceptibility to Ewing sarcoma. Nat Genet 44 (3): 323-7, 2012.
- Machiela MJ, Grünewald TGP, Surdez D, et al.: Genome-wide association study identifies multiple new loci associated with Ewing sarcoma susceptibility. Nat Commun 9 (1): 3184, 2018.
- Leavey PJ, Laack NN, Krailo MD, et al.: Phase III Trial Adding Vincristine-Topotecan-Cyclophosphamide to the Initial Treatment of Patients With Nonmetastatic Ewing Sarcoma: A Children's Oncology Group Report. J Clin Oncol 39 (36): 4029-4038, 2021.
- Brasme JF, Chalumeau M, Oberlin O, et al.: Time to diagnosis of Ewing tumors in children and adolescents is not associated with metastasis or survival: a prospective multicenter study of 436 patients. J Clin Oncol 32 (18): 1935-40, 2014.
- Cash T, McIlvaine E, Krailo MD, et al.: Comparison of clinical features and outcomes in patients with extraskeletal versus skeletal localized Ewing sarcoma: A report from the Children's Oncology Group. Pediatr Blood Cancer 63 (10): 1771-9, 2016.
- Applebaum MA, Worch J, Matthay KK, et al.: Clinical features and outcomes in patients with extraskeletal Ewing sarcoma. Cancer 117 (13): 3027-32, 2011.
- Tal AL, Doshi H, Parkar F, et al.: The Utility of 18FDG PET/CT Versus Bone Scan for Identification of Bone Metastases in a Pediatric Sarcoma Population and a Review of the Literature. J Pediatr Hematol Oncol 43 (2): 52-58, 2021.
- Costelloe CM, Chuang HH, Daw NC: PET/CT of Osteosarcoma and Ewing Sarcoma. Semin Roentgenol 52 (4): 255-268, 2017.
- Saifuddin A, Michelagnoli M, Pressney I: Skip metastases in appendicular Ewing sarcoma: relationship to distant metastases at diagnosis, chemotherapy response and overall survival. Skeletal Radiol 52 (3): 585-591, 2023.
- Campbell KM, Shulman DS, Grier HE, et al.: Role of bone marrow biopsy for staging new patients with Ewing sarcoma: A systematic review. Pediatr Blood Cancer 68 (2): e28807, 2021.
- Cotterill SJ, Ahrens S, Paulussen M, et al.: Prognostic factors in Ewing's tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing's Sarcoma Study Group. J Clin Oncol 18 (17): 3108-14, 2000.
- Miser JS, Krailo MD, Tarbell NJ, et al.: Treatment of metastatic Ewing's sarcoma or primitive neuroectodermal tumor of bone: evaluation of combination ifosfamide and etoposide--a Children's Cancer Group and Pediatric Oncology Group study. J Clin Oncol 22 (14): 2873-6, 2004.
- Rodríguez-Galindo C, Liu T, Krasin MJ, et al.: Analysis of prognostic factors in ewing sarcoma family of tumors: review of St. Jude Children's Research Hospital studies. Cancer 110 (2): 375-84, 2007.
- Karski EE, McIlvaine E, Segal MR, et al.: Identification of Discrete Prognostic Groups in Ewing Sarcoma. Pediatr Blood Cancer 63 (1): 47-53, 2016.
- Paulussen M, Ahrens S, Craft AW, et al.: Ewing's tumors with primary lung metastases: survival analysis of 114 (European Intergroup) Cooperative Ewing's Sarcoma Studies patients. J Clin Oncol 16 (9): 3044-52, 1998.
- Paulussen M, Ahrens S, Burdach S, et al.: Primary metastatic (stage IV) Ewing tumor: survival analysis of 171 patients from the EICESS studies. European Intergroup Cooperative Ewing Sarcoma Studies. Ann Oncol 9 (3): 275-81, 1998.
- Ladenstein R, Pötschger U, Le Deley MC, et al.: Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol 28 (20): 3284-91, 2010.
- Applebaum MA, Goldsby R, Neuhaus J, et al.: Clinical features and outcomes in patients with Ewing sarcoma and regional lymph node involvement. Pediatr Blood Cancer 59 (4): 617-20, 2012.
- Bacci G, Longhi A, Ferrari S, et al.: Prognostic factors in non-metastatic Ewing's sarcoma tumor of bone: an analysis of 579 patients treated at a single institution with adjuvant or neoadjuvant chemotherapy between 1972 and 1998. Acta Oncol 45 (4): 469-75, 2006.
- Wallace MW, Niec JA, Ghani MOA, et al.: Distribution and Surgical Management of Visceral Ewing Sarcoma Among Children and Adolescents. J Pediatr Surg 58 (9): 1727-1735, 2023.
- Ahrens S, Hoffmann C, Jabar S, et al.: Evaluation of prognostic factors in a tumor volume-adapted treatment strategy for localized Ewing sarcoma of bone: the CESS 86 experience. Cooperative Ewing Sarcoma Study. Med Pediatr Oncol 32 (3): 186-95, 1999.
- De Ioris MA, Prete A, Cozza R, et al.: Ewing sarcoma of the bone in children under 6 years of age. PLoS One 8 (1): e53223, 2013.
- Huh WW, Daw NC, Herzog CE, et al.: Ewing sarcoma family of tumors in children younger than 10 years of age. Pediatr Blood Cancer 64 (4): , 2017.
- Ahmed SK, Randall RL, DuBois SG, et al.: Identification of Patients With Localized Ewing Sarcoma at Higher Risk for Local Failure: A Report From the Children's Oncology Group. Int J Radiat Oncol Biol Phys 99 (5): 1286-1294, 2017.
- Grier HE, Krailo MD, Tarbell NJ, et al.: Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 348 (8): 694-701, 2003.
- Granowetter L, Womer R, Devidas M, et al.: Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: a Children's Oncology Group Study. J Clin Oncol 27 (15): 2536-41, 2009.
- Womer RB, West DC, Krailo MD, et al.: Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 30 (33): 4148-54, 2012.
- Pieper S, Ranft A, Braun-Munzinger G, et al.: Ewing's tumors over the age of 40: a retrospective analysis of 47 patients treated according to the International Clinical Trials EICESS 92 and EURO-E.W.I.N.G. 99. Onkologie 31 (12): 657-63, 2008.
- Wong T, Goldsby RE, Wustrack R, et al.: Clinical features and outcomes of infants with Ewing sarcoma under 12 months of age. Pediatr Blood Cancer 62 (11): 1947-51, 2015.
- Worch J, Ranft A, DuBois SG, et al.: Age dependency of primary tumor sites and metastases in patients with Ewing sarcoma. Pediatr Blood Cancer 65 (9): e27251, 2018.
- Schlegel M, Zeumer M, Prodinger PM, et al.: Impact of Pathological Fractures on the Prognosis of Primary Malignant Bone Sarcoma in Children and Adults: A Single-Center Retrospective Study of 205 Patients. Oncology 94 (6): 354-362, 2018.
- Bramer JA, Abudu AA, Grimer RJ, et al.: Do pathological fractures influence survival and local recurrence rate in bony sarcomas? Eur J Cancer 43 (13): 1944-51, 2007.
- Applebaum MA, Goldsby R, Neuhaus J, et al.: Clinical features and outcomes in patients with secondary Ewing sarcoma. Pediatr Blood Cancer 60 (4): 611-5, 2013.
- Roberts P, Burchill SA, Brownhill S, et al.: Ploidy and karyotype complexity are powerful prognostic indicators in the Ewing's sarcoma family of tumors: a study by the United Kingdom Cancer Cytogenetics and the Children's Cancer and Leukaemia Group. Genes Chromosomes Cancer 47 (3): 207-20, 2008.
- Hattinger CM, Pötschger U, Tarkkanen M, et al.: Prognostic impact of chromosomal aberrations in Ewing tumours. Br J Cancer 86 (11): 1763-9, 2002.
- Mackintosh C, Ordóñez JL, García-Domínguez DJ, et al.: 1q gain and CDT2 overexpression underlie an aggressive and highly proliferative form of Ewing sarcoma. Oncogene 31 (10): 1287-98, 2012.
- Tirode F, Surdez D, Ma X, et al.: Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov 4 (11): 1342-53, 2014.
- Mrózek K, Bloomfield CD: Der(16)t(1;16) is a secondary chromosome aberration in at least eighteen different types of human cancer. Genes Chromosomes Cancer 23 (1): 78-80, 1998.
- Mugneret F, Lizard S, Aurias A, et al.: Chromosomes in Ewing's sarcoma. II. Nonrandom additional changes, trisomy 8 and der(16)t(1;16). Cancer Genet Cytogenet 32 (2): 239-45, 1988.
- Hattinger CM, Rumpler S, Ambros IM, et al.: Demonstration of the translocation der(16)t(1;16)(q12;q11.2) in interphase nuclei of Ewing tumors. Genes Chromosomes Cancer 17 (3): 141-50, 1996.
- Dubois SG, Epling CL, Teague J, et al.: Flow cytometric detection of Ewing sarcoma cells in peripheral blood and bone marrow. Pediatr Blood Cancer 54 (1): 13-8, 2010.
- Schleiermacher G, Peter M, Oberlin O, et al.: Increased risk of systemic relapses associated with bone marrow micrometastasis and circulating tumor cells in localized ewing tumor. J Clin Oncol 21 (1): 85-91, 2003.
- Zoubek A, Ladenstein R, Windhager R, et al.: Predictive potential of testing for bone marrow involvement in Ewing tumor patients by RT-PCR: a preliminary evaluation. Int J Cancer 79 (1): 56-60, 1998.
- Shukla NN, Patel JA, Magnan H, et al.: Plasma DNA-based molecular diagnosis, prognostication, and monitoring of patients with EWSR1 fusion-positive sarcomas. JCO Precis Oncol 2017: , 2017.
- Shulman DS, Klega K, Imamovic-Tuco A, et al.: Detection of circulating tumour DNA is associated with inferior outcomes in Ewing sarcoma and osteosarcoma: a report from the Children's Oncology Group. Br J Cancer 119 (5): 615-621, 2018.
- Krumbholz M, Eiblwieser J, Ranft A, et al.: Quantification of Translocation-Specific ctDNA Provides an Integrating Parameter for Early Assessment of Treatment Response and Risk Stratification in Ewing Sarcoma. Clin Cancer Res 27 (21): 5922-5930, 2021.
- Vo KT, Edwards JV, Epling CL, et al.: Impact of Two Measures of Micrometastatic Disease on Clinical Outcomes in Patients with Newly Diagnosed Ewing Sarcoma: A Report from the Children's Oncology Group. Clin Cancer Res 22 (14): 3643-50, 2016.
- Lerman DM, Monument MJ, McIlvaine E, et al.: Tumoral TP53 and/or CDKN2A alterations are not reliable prognostic biomarkers in patients with localized Ewing sarcoma: a report from the Children's Oncology Group. Pediatr Blood Cancer 62 (5): 759-65, 2015.
- Shulman DS, Chen S, Hall D, et al.: Adverse prognostic impact of the loss of STAG2 protein expression in patients with newly diagnosed localised Ewing sarcoma: A report from the Children's Oncology Group. Br J Cancer 127 (12): 2220-2226, 2022.
- Parham DM, Hijazi Y, Steinberg SM, et al.: Neuroectodermal differentiation in Ewing's sarcoma family of tumors does not predict tumor behavior. Hum Pathol 30 (8): 911-8, 1999.
- Luksch R, Sampietro G, Collini P, et al.: Prognostic value of clinicopathologic characteristics including neuroectodermal differentiation in osseous Ewing's sarcoma family of tumors in children. Tumori 85 (2): 101-7, 1999 Mar-Apr.
- de Alava E, Kawai A, Healey JH, et al.: EWS-FLI1 fusion transcript structure is an independent determinant of prognosis in Ewing's sarcoma. J Clin Oncol 16 (4): 1248-55, 1998.
- van Doorninck JA, Ji L, Schaub B, et al.: Current treatment protocols have eliminated the prognostic advantage of type 1 fusions in Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 28 (12): 1989-94, 2010.
- Le Deley MC, Delattre O, Schaefer KL, et al.: Impact of EWS-ETS fusion type on disease progression in Ewing's sarcoma/peripheral primitive neuroectodermal tumor: prospective results from the cooperative Euro-E.W.I.N.G. 99 trial. J Clin Oncol 28 (12): 1982-8, 2010.
- Paulussen M, Ahrens S, Dunst J, et al.: Localized Ewing tumor of bone: final results of the cooperative Ewing's Sarcoma Study CESS 86. J Clin Oncol 19 (6): 1818-29, 2001.
- Rosito P, Mancini AF, Rondelli R, et al.: Italian Cooperative Study for the treatment of children and young adults with localized Ewing sarcoma of bone: a preliminary report of 6 years of experience. Cancer 86 (3): 421-8, 1999.
- Wunder JS, Paulian G, Huvos AG, et al.: The histological response to chemotherapy as a predictor of the oncological outcome of operative treatment of Ewing sarcoma. J Bone Joint Surg Am 80 (7): 1020-33, 1998.
- Oberlin O, Deley MC, Bui BN, et al.: Prognostic factors in localized Ewing's tumours and peripheral neuroectodermal tumours: the third study of the French Society of Paediatric Oncology (EW88 study). Br J Cancer 85 (11): 1646-54, 2001.
- Lozano-Calderón SA, Albergo JI, Groot OQ, et al.: Complete tumor necrosis after neoadjuvant chemotherapy defines good responders in patients with Ewing sarcoma. Cancer 129 (1): 60-70, 2023.
- Ferrari S, Bertoni F, Palmerini E, et al.: Predictive factors of histologic response to primary chemotherapy in patients with Ewing sarcoma. J Pediatr Hematol Oncol 29 (6): 364-8, 2007.
- Hawkins DS, Schuetze SM, Butrynski JE, et al.: [18F]Fluorodeoxyglucose positron emission tomography predicts outcome for Ewing sarcoma family of tumors. J Clin Oncol 23 (34): 8828-34, 2005.
- Denecke T, Hundsdörfer P, Misch D, et al.: Assessment of histological response of paediatric bone sarcomas using FDG PET in comparison to morphological volume measurement and standardized MRI parameters. Eur J Nucl Med Mol Imaging 37 (10): 1842-53, 2010.
- Palmerini E, Colangeli M, Nanni C, et al.: The role of FDG PET/CT in patients treated with neoadjuvant chemotherapy for localized bone sarcomas. Eur J Nucl Med Mol Imaging 44 (2): 215-223, 2017.
- Lin PP, Jaffe N, Herzog CE, et al.: Chemotherapy response is an important predictor of local recurrence in Ewing sarcoma. Cancer 109 (3): 603-11, 2007.
- Bosma SE, Rueten-Budde AJ, Lancia C, et al.: Individual risk evaluation for local recurrence and distant metastasis in Ewing sarcoma: A multistate model: A multistate model for Ewing sarcoma. Pediatr Blood Cancer 66 (11): e27943, 2019.
- Reiter AJ, Huang L, Craig BT, et al.: Survival outcomes in pediatric patients with metastatic Ewing sarcoma who achieve a rapid complete response of pulmonary metastases. Pediatr Blood Cancer 71 (7): e31026, 2024.
Cellular Classification of Ewing Sarcoma
Ewing sarcoma belongs to the group of neoplasms commonly referred to as small round blue cell tumors of childhood. The individual cells of Ewing sarcoma contain round-to-oval nuclei, with fine dispersed chromatin without nucleoli. Occasionally, cells with smaller, more hyperchromatic, and probably degenerative nuclei are present, giving a light cell/dark cell pattern. The cytoplasm varies in amount, but in the classic case, it is clear and contains glycogen, which can be highlighted with a periodic acid-Schiff stain. The tumor cells are tightly packed and grow in a diffuse pattern without evidence of structural organization. Tumors with the requisite translocation that show neuronal differentiation are not considered a separate entity, but rather, part of a continuum of differentiation.
CD99 is a surface membrane protein that is expressed in most cases of Ewing sarcoma and is useful in diagnosing these tumors when the results are interpreted in the context of clinical and pathological parameters.[1] CD99 positivity is not unique to Ewing sarcoma, and positivity by immunochemistry is found in several other tumors, including synovial sarcoma, non-Hodgkin lymphoma, and gastrointestinal stromal tumors. NKX2.2 is a nuclear antigen that is also commonly assessed by immunohistochemistry to support a diagnosis of Ewing sarcoma, although it is also not 100% specific for this diagnosis.[2]
For more information about the cellular classification of other undifferentiated small round cell sarcomas, see the Undifferentiated Small Round Cell (Ewing-Like) Sarcomas section.
References:
- Parham DM, Hijazi Y, Steinberg SM, et al.: Neuroectodermal differentiation in Ewing's sarcoma family of tumors does not predict tumor behavior. Hum Pathol 30 (8): 911-8, 1999.
- Yoshida A, Sekine S, Tsuta K, et al.: NKX2.2 is a useful immunohistochemical marker for Ewing sarcoma. Am J Surg Pathol 36 (7): 993-9, 2012.
Genomics of Ewing Sarcoma
Molecular Features of Ewing Sarcoma
The World Health Organization identifies the presence of a gene fusion involving EWSR1 or FUS and a gene in the ETS family as a defining element of Ewing sarcoma.[1] The EWSR1 gene located on chromosome 22 band q12 is a member of the FET family (FUS, EWSR1, TAF15) of RNA-binding proteins.[2] Characteristically, the amino terminus of the EWSR1 gene is juxtaposed with the carboxy terminus of a gene from the ETS family of DNA-binding transcription factors (see Table 4). The FLI1 gene located on chromosome 11 band q24 is a member of the ETS family and is the ETS family fusion partner for EWSR1 in 85% to 90% of pediatric cases of Ewing sarcoma.[3,4,5] Other ETS family members that may combine with the EWSR1 gene are ERG, ETV1, ETV4, and FEV.[6] Rarely, FUS, another FET family member, can substitute for EWSR1.[7] Finally, there are a few rare cases in which EWSR1 has translocated with partners that are not members of the ETS family of oncogenes. These tumors are thought to be distinct from Ewing sarcoma and are discussed separately. For more information, see the Undifferentiated Small Round Cell (Ewing-Like) Sarcomas section.
The EWSR1::FLI1 translocation associated with Ewing sarcoma can occur at several potential breakpoints in each of the genes that join to form the novel segment of DNA. Once thought to be significant,[8] two large series have shown that the EWSR1::FLI1 translocation breakpoint site is not an adverse prognostic factor.[9,10]
Besides the consistent aberrations involving the EWSR1 gene, secondary numerical and structural chromosomal aberrations are observed in most cases of Ewing sarcoma. Chromosome gains are more common than chromosome losses, and structural chromosome imbalances are also observed.[11] Two of the more common chromosome aberrations are those involving chromosome 8 or chromosomes 1 and 16.[11]
- Gain of whole chromosome 8 (trisomy 8). Trisomy 8 is the most frequent chromosomal alteration in Ewing sarcoma, occurring in nearly 50% of tumors.[3,4] Gain of chromosome 8 does not appear to have prognostic significance.[3,12]
- Gain of chromosome 1q and loss of chromosome 16q. These occur in approximately 20% of patients and often occur together. Gain of chromosome 1q and/or deletion of chromosome 16q has been associated with inferior prognosis for patients with Ewing sarcoma in several cohorts.[3,13,14] These two chromosomal alterations commonly occur together across a range of cancer types, including Ewing sarcoma.[15] Their co-occurrence is likely a result of their derivation from an unbalanced t(1;16) translocation resulting in gain of chromosome 1q together with loss of chromosomal material from 16q.[12,16]
The genomic landscape of Ewing sarcoma is characterized by a relatively silent genome, with a paucity of variants in pathways that might be amenable to treatment with novel targeted therapies.[3,4,5] Recurring genomic alterations are described below. For some of these genomic alterations, claims of prognostic significance have been made. However, these claims need to be viewed cautiously because of the relatively small size of most studies, the low frequency of many of the genomic alterations, the variable use of tumor tissue from diagnosis versus relapse specimens, and the need to consider clinical prognostic factors such as tumor size and the presence of metastatic disease.
- STAG2 variants. Variants in STAG2, a member of the cohesin complex, occur in about 15% to 20% of the cases.[3,4,5] These variants lead to loss of STAG2 expression and function in tumor cells.[5] Loss of STAG2 expression (detected by immunohistochemistry [IHC]) has been observed in tumors in which a STAG2 variant cannot be detected. In one report, loss of STAG2 expression by IHC was associated with inferior prognosis.[17]
- CDKN2A deletions. CDKN2A deletions have been noted in 12% to 22% of cases.[3,4,5]
- TP53 variants. TP53 variants were identified in about 6% to 7% of Ewing sarcoma cases reported by pediatric research teams.[3,4,5] Higher rates of TP53 variants (up to 19%) have been described in cohorts from single institutions that contain higher proportions of adult patients.[18,19] The coexistence of STAG2 and TP53 variants has been associated with a poor clinical outcome in one retrospective report.
- ERF alterations. Genomic alterations in ERF leading to loss of function (frameshift, missense, and deep deletion) were reported in 7% of Ewing sarcoma tumors.[18] A second report observed ERF alterations at a rate of 3% in another Ewing sarcoma cohort.[4]
- Other genes with recurring genomic alterations in Ewing sarcoma. Recurring genomic alterations present in fewer than 5% of Ewing sarcoma patients were reported: EZH2,[3,19]BCOR,[3]SMARCA4,[19]CREBBP,[19]TERT, and FGFR1.[18]
Ewing sarcoma translocations can all be found with standard cytogenetic analysis. A fluorescence in situ hybridization (FISH) rapid analysis looking for a break apart of the EWSR1 gene is now frequently done to confirm the diagnosis of Ewing sarcoma molecularly.[20] This test result must be considered with caution, however. Ewing sarcomas that harbor FUS translocations will have negative tests because the EWSR1 gene is not translocated in those cases. In addition, other small round tumors also contain translocations of different ETS family members with EWSR1, such as desmoplastic small round cell tumor, clear cell sarcoma, extraskeletal myxoid chondrosarcoma, and myxoid liposarcoma, all of which may be positive with a EWSR1 FISH break-apart probe. A detailed analysis of 85 patients with small round blue cell tumors that were negative for EWSR1 rearrangement by FISH (with an EWSR1 break-apart probe) identified eight patients with FUS rearrangements.[21] Four patients who had EWSR1::ERG fusions were not detected by FISH with an EWSR1 break-apart probe. The authors do not recommend relying solely on EWSR1 break-apart probes for analyzing small round blue cell tumors with strong immunohistochemical positivity for CD99. Next-generation sequencing assays, including dedicated fusion panels, are now commonly used in the evaluation of these tumors.
Table 4.EWSR1andFUSFusions and Translocations in Ewing Sarcoma| FET Family Partner | Fusion With ETS-Like Oncogene Partner | Translocation | Comment |
|---|
| a These partners are not members of the ETS family of oncogenes; therefore, these tumors are not classified as Ewing sarcoma. |
| EWSR1 | EWSR1::FLI1 | t(11;22)(q24;q12) | Most common; approximately 85% to 90% of cases |
| EWSR1::ERG | t(21;22)(q22;q12) | Second most common; approximately 10% of cases |
| EWSR1::ETV1 | t(7;22)(p22;q12) | Rare |
| EWSR1::ETV4 | t(17;22)(q12;q12) | Rare |
| EWSR1::FEV | t(2;22)(q35;q12) | Rare |
| EWSR1::NFATC2a | t(20;22)(q13;q12) | Rare |
| EWSR1::POU5F1a | t(6;22)(p21;q12) | |
| EWSR1::SMARCA5a | t(4;22)(q31;q12) | Rare |
| EWSR1::PATZ1a | t(6;22)(p21;q12) | |
| EWSR1::SP3a | t(2;22)(q31;q12) | Rare |
| FUS | FUS::ERG | t(16;21)(p11;q22) | Rare |
| FUS::FEV | t(2;16)(q35;p11) | Rare |
References:
- WHO Classification of Tumours Editorial Board: WHO Classification of Tumours. Volume 3: Soft Tissue and Bone Tumours. 5th ed., IARC Press, 2020.
- Schwartz JC, Cech TR, Parker RR: Biochemical Properties and Biological Functions of FET Proteins. Annu Rev Biochem 84: 355-79, 2015.
- Tirode F, Surdez D, Ma X, et al.: Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov 4 (11): 1342-53, 2014.
- Crompton BD, Stewart C, Taylor-Weiner A, et al.: The genomic landscape of pediatric Ewing sarcoma. Cancer Discov 4 (11): 1326-41, 2014.
- Brohl AS, Solomon DA, Chang W, et al.: The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet 10 (7): e1004475, 2014.
- Hattinger CM, Rumpler S, Strehl S, et al.: Prognostic impact of deletions at 1p36 and numerical aberrations in Ewing tumors. Genes Chromosomes Cancer 24 (3): 243-54, 1999.
- Sankar S, Lessnick SL: Promiscuous partnerships in Ewing's sarcoma. Cancer Genet 204 (7): 351-65, 2011.
- de Alava E, Kawai A, Healey JH, et al.: EWS-FLI1 fusion transcript structure is an independent determinant of prognosis in Ewing's sarcoma. J Clin Oncol 16 (4): 1248-55, 1998.
- van Doorninck JA, Ji L, Schaub B, et al.: Current treatment protocols have eliminated the prognostic advantage of type 1 fusions in Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 28 (12): 1989-94, 2010.
- Le Deley MC, Delattre O, Schaefer KL, et al.: Impact of EWS-ETS fusion type on disease progression in Ewing's sarcoma/peripheral primitive neuroectodermal tumor: prospective results from the cooperative Euro-E.W.I.N.G. 99 trial. J Clin Oncol 28 (12): 1982-8, 2010.
- Roberts P, Burchill SA, Brownhill S, et al.: Ploidy and karyotype complexity are powerful prognostic indicators in the Ewing's sarcoma family of tumors: a study by the United Kingdom Cancer Cytogenetics and the Children's Cancer and Leukaemia Group. Genes Chromosomes Cancer 47 (3): 207-20, 2008.
- Hattinger CM, Rumpler S, Ambros IM, et al.: Demonstration of the translocation der(16)t(1;16)(q12;q11.2) in interphase nuclei of Ewing tumors. Genes Chromosomes Cancer 17 (3): 141-50, 1996.
- Hattinger CM, Pötschger U, Tarkkanen M, et al.: Prognostic impact of chromosomal aberrations in Ewing tumours. Br J Cancer 86 (11): 1763-9, 2002.
- Mackintosh C, Ordóñez JL, García-Domínguez DJ, et al.: 1q gain and CDT2 overexpression underlie an aggressive and highly proliferative form of Ewing sarcoma. Oncogene 31 (10): 1287-98, 2012.
- Mrózek K, Bloomfield CD: Der(16)t(1;16) is a secondary chromosome aberration in at least eighteen different types of human cancer. Genes Chromosomes Cancer 23 (1): 78-80, 1998.
- Mugneret F, Lizard S, Aurias A, et al.: Chromosomes in Ewing's sarcoma. II. Nonrandom additional changes, trisomy 8 and der(16)t(1;16). Cancer Genet Cytogenet 32 (2): 239-45, 1988.
- Shulman DS, Chen S, Hall D, et al.: Adverse prognostic impact of the loss of STAG2 protein expression in patients with newly diagnosed localised Ewing sarcoma: A report from the Children's Oncology Group. Br J Cancer 127 (12): 2220-2226, 2022.
- Ogura K, Elkrief A, Bowman AS, et al.: Prospective Clinical Genomic Profiling of Ewing Sarcoma: ERF and FGFR1 Mutations as Recurrent Secondary Alterations of Potential Biologic and Therapeutic Relevance. JCO Precis Oncol 6: e2200048, 2022.
- Rock A, Uche A, Yoon J, et al.: Bioinformatic Analysis of Recurrent Genomic Alterations and Corresponding Pathway Alterations in Ewing Sarcoma. J Pers Med 13 (10): , 2023.
- Monforte-Muñoz H, Lopez-Terrada D, Affendie H, et al.: Documentation of EWS gene rearrangements by fluorescence in-situ hybridization (FISH) in frozen sections of Ewing's sarcoma-peripheral primitive neuroectodermal tumor. Am J Surg Pathol 23 (3): 309-15, 1999.
- Chen S, Deniz K, Sung YS, et al.: Ewing sarcoma with ERG gene rearrangements: A molecular study focusing on the prevalence of FUS-ERG and common pitfalls in detecting EWSR1-ERG fusions by FISH. Genes Chromosomes Cancer 55 (4): 340-9, 2016.
Stage Information for Ewing Sarcoma
Pretreatment staging studies for Ewing sarcoma may include the following:
- Magnetic resonance imaging (MRI) of the primary site.
- Computed tomography (CT) scan of the primary site and chest.
- Positron emission tomography using fluorine F 18-fludeoxyglucose (18F-FDG PET) or 18F-FDG PET-CT.
- Bone scan has traditionally been part of the staging evaluation for Ewing sarcoma. However, many investigators believe that PET scan can replace bone scan.[1,2]
- Bone marrow aspiration and biopsy.
For patients with confirmed Ewing sarcoma, pretreatment staging studies include MRI and/or CT scan, depending on the primary site. Despite the fact that CT and MRI are both equivalent in terms of staging, use of both imaging modalities may help radiation therapy planning.[3] Whole-body MRI may provide additional information that could potentially alter therapy planning.[4] Additional pretreatment staging studies include bone scan and CT scan of the chest. In certain studies, determination of pretreatment tumor volume is an important variable.
18F-FDG PET-CT scans have demonstrated high sensitivity and specificity in Ewing sarcoma and are now routinely used to complete staging. In one institutional study, 18F-FDG PET had a very high correlation with bone scan; the investigators suggested that it could replace bone scan for the initial extent of disease evaluation.[5] This finding was confirmed in a single-institution retrospective review.[6] 18F-FDG PET-CT is more accurate than 18F-FDG PET alone in Ewing sarcoma.[7,8,9]
Bone marrow aspiration and biopsy have been considered the standard of care for Ewing sarcoma. However, two retrospective studies showed that for patients (N = 141) who were evaluated by bone scan and/or PET scan and lung CT without evidence of metastases, bone marrow aspirates and biopsies were negative in every case.[5,10] A single-institution retrospective review of 504 patients with Ewing sarcoma identified 12 patients with bone marrow metastasis.[11] Only one patient was found to have bone marrow involvement without any other sites of metastatic disease, for an incidence of 1 per 367 (0.3%) in patients with clinically localized disease. The need for routine use of bone marrow aspirates and biopsies in patients without bone metastases is now in question.
For Ewing sarcoma, tumors are practically staged as localized or metastatic, and other staging systems are not commonly used. The tumor is defined as localized when, by clinical and imaging techniques, there is no spread beyond the primary site or regional lymph node involvement. Continuous extension into adjacent soft tissue may occur. If there is a question of regional lymph node involvement, pathological confirmation is indicated.
References:
- Costelloe CM, Chuang HH, Daw NC: PET/CT of Osteosarcoma and Ewing Sarcoma. Semin Roentgenol 52 (4): 255-268, 2017.
- Tal AL, Doshi H, Parkar F, et al.: The Utility of 18FDG PET/CT Versus Bone Scan for Identification of Bone Metastases in a Pediatric Sarcoma Population and a Review of the Literature. J Pediatr Hematol Oncol 43 (2): 52-58, 2021.
- Meyer JS, Nadel HR, Marina N, et al.: Imaging guidelines for children with Ewing sarcoma and osteosarcoma: a report from the Children's Oncology Group Bone Tumor Committee. Pediatr Blood Cancer 51 (2): 163-70, 2008.
- Mentzel HJ, Kentouche K, Sauner D, et al.: Comparison of whole-body STIR-MRI and 99mTc-methylene-diphosphonate scintigraphy in children with suspected multifocal bone lesions. Eur Radiol 14 (12): 2297-302, 2004.
- Newman EN, Jones RL, Hawkins DS: An evaluation of [F-18]-fluorodeoxy-D-glucose positron emission tomography, bone scan, and bone marrow aspiration/biopsy as staging investigations in Ewing sarcoma. Pediatr Blood Cancer 60 (7): 1113-7, 2013.
- Ulaner GA, Magnan H, Healey JH, et al.: Is methylene diphosphonate bone scan necessary for initial staging of Ewing sarcoma if 18F-FDG PET/CT is performed? AJR Am J Roentgenol 202 (4): 859-67, 2014.
- Völker T, Denecke T, Steffen I, et al.: Positron emission tomography for staging of pediatric sarcoma patients: results of a prospective multicenter trial. J Clin Oncol 25 (34): 5435-41, 2007.
- Gerth HU, Juergens KU, Dirksen U, et al.: Significant benefit of multimodal imaging: PET/CT compared with PET alone in staging and follow-up of patients with Ewing tumors. J Nucl Med 48 (12): 1932-9, 2007.
- Treglia G, Salsano M, Stefanelli A, et al.: Diagnostic accuracy of ¹⁸F-FDG-PET and PET/CT in patients with Ewing sarcoma family tumours: a systematic review and a meta-analysis. Skeletal Radiol 41 (3): 249-56, 2012.
- Kopp LM, Hu C, Rozo B, et al.: Utility of bone marrow aspiration and biopsy in initial staging of Ewing sarcoma. Pediatr Blood Cancer 62 (1): 12-5, 2015.
- Cesari M, Righi A, Colangeli M, et al.: Bone marrow biopsy in the initial staging of Ewing sarcoma: Experience from a single institution. Pediatr Blood Cancer 66 (6): e27653, 2019.
Treatment Option Overview for Ewing Sarcoma
It is important that patients be evaluated by specialists from the appropriate disciplines (e.g., medical oncologists, surgical or orthopedic oncologists, and radiation oncologists) as early as possible. Multidisciplinary review with radiologists and pathologists is often performed at sarcoma specialty centers.
Appropriate imaging studies of the suspected primary site are obtained before biopsy. To ensure that the biopsy incision is placed in an acceptable location, the surgical or orthopedic oncologist (who will perform the definitive surgery) is consulted on biopsy-incision placement. This is especially important if it is thought that the lesion can subsequently be totally excised after initial systemic therapy or if a limb salvage procedure may be attempted. It is almost never appropriate to attempt a primary resection of known Ewing sarcoma at initial diagnosis. With rare exceptions, Ewing sarcoma is sensitive to chemotherapy and will respond to initial systemic therapy. This therapy reduces the risk of tumor spread to surrounding tissues and makes ultimate surgery easier and safer. Biopsy should be from soft tissue as often as possible to avoid increasing the risk of fracture.[1] If the initial biopsy sample is obtained from bone, reserving some tissue without decalcification is required because decalcification denatures DNA and makes genomic profiling of tumor tissue impossible.[2] The pathologist is consulted before biopsy/surgery to ensure that the incision will not compromise the radiation port and that multiple types of adequate tissue samples are obtained. It is important to obtain fresh tissue, whenever possible, for cytogenetics and molecular pathology. A second option is to perform a needle biopsy, as long as adequate tissue is obtained for molecular studies.[3]
Table 5 describes the treatment options for localized, metastatic, and recurrent Ewing sarcoma.
The successful treatment of patients with Ewing sarcoma requires systemic chemotherapy [4,5,6,7,8,9,10] in conjunction with surgery and/or radiation therapy for local tumor control.[11,12,13,14,15] In general, patients receive chemotherapy before instituting local-control measures. In patients who undergo surgery, surgical margins and histological response are considered in planning postoperative therapy. Patients with metastatic disease often have a good initial response to preoperative chemotherapy, but in most cases, the disease is only partially controlled or recurs.[16,17,18,19,20,21] Patients with lung as the only metastatic site have a better prognosis than do patients with metastases to bone and/or bone marrow. Adequate local control for metastatic sites, particularly bone metastases, may be an important consideration.[22]
Chemotherapy for Ewing Sarcoma
Multidrug chemotherapy for Ewing sarcoma always includes vincristine, doxorubicin, ifosfamide, and etoposide. Most protocols also use cyclophosphamide and some incorporate dactinomycin. The mode of administration and dose intensity of cyclophosphamide within courses differs markedly between protocols. A European Intergroup Cooperative Ewing Sarcoma Study (EICESS) trial suggested that 1.2 g of cyclophosphamide produced a similar event-free survival (EFS) compared with 6 g of ifosfamide in patients with lower-risk disease. The trial also identified a trend toward better EFS for patients with localized Ewing sarcoma and higher-risk disease when treatment included etoposide (GER-GPOH-EICESS-92 [NCT00002516]).[23][Level of evidence A1]
Protocols in the United States generally alternate courses of vincristine, cyclophosphamide, and doxorubicin (VDC) with courses of ifosfamide and etoposide (IE),[8] using interval compression.[24,25,26] For many years, European protocols generally combined vincristine, doxorubicin, and an alkylating agent with or without etoposide in a single treatment cycle.[10] After the completion of the randomized EURO EWING 2012 (EE2012) trial (see below), European investigators shifted to therapy with cycles of VDC alternating with cycles of IE.[27][Level of evidence B1] The duration of primary chemotherapy ranges from 6 months to approximately 1 year.
Evidence (chemotherapy):
- An international consortium of European countries conducted the EURO-EWING-INTERGROUP-EE99 (NCT00020566) trial from 2000 to 2010.[28][Level of evidence A1] All patients received induction therapy with six cycles of vincristine, ifosfamide, doxorubicin, and etoposide (VIDE), followed by local control, and then one cycle of vincristine, dactinomycin, and ifosfamide (VAI). Patients were classified as standard risk if they had localized disease and good histological response to therapy or if they had localized tumors less than 200 mL in volume at presentation; they were treated with radiation therapy alone as local treatment. Standard-risk patients (n = 856) were randomly assigned to receive maintenance therapy with either seven cycles of vincristine, dactinomycin, and cyclophosphamide (VAC) or VAI.
- There was no significant difference in EFS or overall survival (OS) between patients who received VAC and patients who received VAI.
- The 3-year EFS rate for this low-risk population was 77%.
- It is difficult to compare this outcome with that of other large series because the study population excluded patients with poor response to initial therapy or patients with tumors more than 200 mL in volume who received local-control therapy with radiation alone. All other published series report results for all patients who present without clinically detectable metastasis; thus, these other series included patients with poor response and patients with larger primary tumors treated with radiation alone, all of whom were excluded from the EURO-EWING-INTERGROUP-EE99 study.
- In a Children's Oncology Group (COG) study (COG-AEWS0031 [NCT00006734]), patients presenting without metastases were randomly assigned to receive cycles of VDC alternating with cycles of IE at either 2-week or 3-week intervals.[24]
- The administration of cycles of VDC/IE at 2-week intervals achieved superior EFS (5-year EFS rate, 73%) than did alternating cycles at 3-week intervals (5-year EFS rate, 65%). With longer follow-up, the advantage of interval-compressed chemotherapy was confirmed.[25]
- The 10-year EFS rate was 70% using interval-compressed chemotherapy, compared with 61% using standard-timing chemotherapy (P = .03). The 10-year OS rate was 76% using interval-compressed chemotherapy, compared with 69% using standard-timing chemotherapy (P = .04).
- The EE2012 trial was an international multicenter phase III study that included two randomized treatments, the European VIDE induction regimen and the North American standard VDC/IE induction regimen. Patients with both localized and metastatic Ewing sarcoma were eligible for the study.[27][Level of evidence B1]
- The hazard ratios (HRs) for EFS (0.71) and OS (0.62) favored VDC/IE over VIDE. The posterior probabilities were 99% for both EFS and OS, which showed that VDC/IE was superior.
- Rates of febrile neutropenia were higher with the VIDE regimen. There were no other major differences in acute toxicities between the two regimens.
- The benefit of VDC/IE over VIDE was seen across subgroups defined by patient age, sex, stage, tumor volume, or country of residence.
- The Brazilian Cooperative Study Group performed a multi-institutional trial that incorporated carboplatin into a risk-adapted intensive regimen in 175 children with localized or metastatic Ewing sarcoma.[29][Level of evidence B4]
- The investigators found significantly increased toxicity without an improvement in outcome with the addition of carboplatin.
- The COG performed a prospective randomized trial in patients with localized Ewing sarcoma. All patients received cycles of VDC and cycles of IE. Patients were then randomly assigned to receive or not receive additional experimental cycles of vincristine, cyclophosphamide, and topotecan.[26]
- The 5-year EFS rate was 78% (95% confidence interval [CI], 72%–82%) for those treated with experimental therapy and 79% (95% CI, 74%–83%) for patients treated with standard therapy.
- The experimental therapy did not significantly reduce the risk of events (EFS HR for experimental arm vs. standard arm, 0.86; 1-sided P = .19).
Local Control (Surgery and Radiation Therapy) for Ewing Sarcoma
Treatment approaches for Ewing sarcoma and therapeutic aggressiveness must be adjusted to maximize local control while also minimizing morbidity.
Surgery is the most commonly used form of local control.[30] Radiation therapy is an effective alternative modality for local control in cases where the functional or cosmetic morbidity of surgery is deemed too high by experienced surgical oncologists. However, in the immature skeleton, radiation therapy can cause subsequent deformities that may be more morbid than deformities from surgery. When complete surgical resection with pathologically negative margins is not anticipated, surgery is not typically performed, and definitive radiation is used instead. When pathologically positive margins are found, then postoperative radiation therapy is indicated. A multidisciplinary discussion between the experienced radiation oncologist and the surgeon is necessary to determine the best treatment options for local control for a given case. For some marginally resectable lesions, a combined approach of preoperative radiation therapy followed by resection can be used.
Timing of local control may impact outcome. A retrospective review from the National Cancer Database identified 1,318 patients with Ewing sarcoma.[31] Patients who initiated local therapy at 6 to 15 weeks had a 5-year OS rate of 78.7% and a 10-year OS rate of 70.3%, and patients who initiated local therapy after 16 weeks had a 5-year OS rate of 70.4% and a 10-year OS rate of 57.1% (P < .001). The difference in OS according to time to local therapy was more important in patients who received radiation therapy alone.
For patients with metastatic Ewing sarcoma, any benefit of combined surgery and radiation therapy compared with either therapy alone for local control is relatively less substantial because the overall prognosis of these patients is much worse than the prognosis of patients who have localized disease.
Randomized trials that directly compare surgery and radiation therapy do not exist, and their relative roles remain controversial. Although retrospective institutional series suggest better local control and survival with surgery than with radiation therapy, most of these studies are compromised by selection bias. An analysis using propensity scoring to adjust for clinical features that may influence the preference for surgery only, radiation only, or combined surgery and radiation demonstrated that similar EFS is achieved with each mode of local therapy.[30] Data for patients with pelvic primary Ewing sarcoma from a North American intergroup trial showed no difference in local control or survival based on local-control modality—surgery alone, radiation therapy alone, or surgery plus radiation therapy.[32]
The EURO-EWING-INTERGROUP-EE99 (NCT00020566) trial prospectively treated 180 patients with pelvic primary tumors without clinically detectable metastatic disease.[33][Level of evidence B4] A retrospective analysis of outcomes for these patients showed improved survival for patients whose tumors were treated with combined radiation therapy and surgery. The study did not prospectively define criteria for the selection of local-control modalities, and the investigators did not have access to information that would allow them to clarify how decisions for local-control modalities were made. In nonsacral tumors, combined local treatment was associated with a lower local recurrence probability (14% [95% CI, 5%–23%] vs. 33% [95% CI, 19%–47%] at 5 years; P = .015) and a higher OS probability (72% [95% CI, 61%–83%] vs. 47% [95% CI, 33%–62%] at 5 years; P = .024), compared with surgery alone. Even in a subgroup of patients with wide surgical margins and a good histological response to induction treatment, the combined local treatment was associated with a higher OS probability (87% [95% CI, 74%–100%] vs. 51% [95% CI, 33%–69%] at 5 years; P = .009), compared with surgery alone. In patients with bone tumors who underwent surgical treatment—after controlling for tumor site in the pelvis, tumor volume, and surgical margin status—those who did not undergo complete removal of the affected bone (HR, 5.04; 95% CI, 2.07–12.24; P < .001), those with a poor histological response to induction chemotherapy (HR, 3.72; 95% CI, 1.51–9.21; P = .004), and those who did not receive additional radiation therapy (HR, 4.34; 95% CI, 1.71–11.05; P = .002) had a higher risk of death.
For patients who undergo gross-total resection with microscopic residual disease, a radiation therapy dose of 50.4 Gy is recommended. For patients treated with primary radiation therapy, the radiation dose is 55.8 Gy (45 Gy to the initial tumor volume and an additional 10.8 Gy to the postchemotherapy volume).[14,34]
Evidence (postoperative radiation therapy):
- Investigators from St. Jude Children's Research Hospital reported 39 patients with localized Ewing sarcoma who received both surgery and radiation.[14]
- The local failure rate for patients with positive margins was 17%, and the OS rate was 71%.
- The local failure rate for patients with negative margins was 5%, and the OS rate was 94%.
- In a large retrospective Italian study, 45 Gy of adjuvant radiation therapy for patients with inadequate margins did not appear to improve either local control or disease-free survival (DFS).[15]
- These investigators concluded that patients who are anticipated to have suboptimal surgery should be considered for definitive radiation therapy.
- The EURO-EWING-INTERGROUP-EE99 (NCT00020566) study reported the outcomes of 599 patients who presented with localized disease and had surgical resection after initial chemotherapy with at least 90% necrosis of the primary tumor.[34][Level of evidence C2] The protocol recommended postoperative radiation therapy for patients with inadequate surgical margins, vertebral primary tumors, or thoracic tumors with pleural effusion, but the decision to use postoperative radiation therapy was left to the institutional investigator.
- Patients who received postoperative radiation therapy (n = 142) had a lower risk of failure than patients who did not receive postoperative radiation therapy, even after controlling for known prognostic factors, including age, sex, tumor site, clinical response, quality of resection, and histological necrosis. Most of the improvement was seen in a decreased risk of local recurrence. The improvement was greater in patients who had large tumors (>200 mL) and were assessed to have 100% necrosis than in patients who were assessed to have 90% to 100% necrosis.
- There is a clear interaction between systemic therapy and local-control modalities for both local control and DFS. The induction regimen used in the EURO-EWING-INTERGROUP-EE99 study is less intense than the induction regimen used in contemporaneous protocols in the COG, and it is not appropriate to extrapolate the results from the EURO-EWING-INTERGROUP-EE99 study to different systemic chemotherapy regimens.
Thoracic primary tumors
Evidence (surgery):
- The treatment and outcomes for 62 patients with thoracic Ewing sarcoma were reported from the Cooperative Weichteilsarkom Studiengruppe CWS-81, -86, -91, -96, and -2002P trials.[35]
- The 5-year OS rate was 58.7% (95% CI, 52.7%–64.7%), and the EFS rate was 52.8% (95% CI, 46.8%–58.8%).
- Patients with intrathoracic tumors (n = 24) had a worse outcome (EFS rate, 37.5% [95% CI, 27.5%–37.5%]) than patients with chest wall tumors (n = 38; EFS rate, 62.3% [95% CI, 54.3%–70.3%]; P = .008).
- Patients aged 10 years and younger (n = 38) had a better survival (EFS rate, 65.7% [95% CI, 57.7%–73.7%]) than patients older than 10 years (EFS rate, 31.3% [95% CI, 21.3%–41.3%]; P = .01).
- Tumor size of less than or equal to 5 cm (n = 15) was associated with significantly better survival (EFS rate, 93.3% [95% CI, 87.3%–99.3%]), compared with a tumor size greater than 5 cm (n = 47; EFS rate, 40% [95% CI, 33%–47%]; P = .002).
- Primary resections were carried out in 36 patients, 75% of which were incomplete, resulting in inferior EFS (P = .006).
- Complete secondary resections were performed in 22 of 40 patients.
- The COG reviewed its results for 98 patients with chest wall tumors who were treated on the INT-0091 and INT-0154 trials from 1988 to 1998 and found the following:[36]
- The 5-year EFS rate was 56%.
- Negative margins were more common in patients who received initial chemotherapy and then underwent resections (41 of 53 patients, 77%) than in patients who had up-front surgery (10 of 20 patients, 50%).
- More patients who underwent up-front surgery received radiation therapy (71%) than patients who started with chemotherapy (48%).
In summary, surgery is chosen as definitive local therapy for suitable patients, but radiation therapy is appropriate for patients with unresectable disease or those who would experience functional or cosmetic compromise by definitive surgery. The possibility of impaired function or cosmesis needs to be measured against the possibility of second tumors in the radiation field. Adjuvant radiation therapy should be considered for patients with residual microscopic disease or inadequate margins.
When preoperative assessment has suggested a high probability that surgical margins will be close or positive, preoperative radiation therapy has achieved tumor shrinkage and allowed surgical resection with clear margins.[37]
Multiple analyses have evaluated diagnostic findings, treatment, and outcome of patients with bone lesions at the following anatomical primary sites:
- Pelvis.[38,39,40]
- Femur.[41,42]
- Humerus.[43,44]
- Hand and foot.[45,46]
- Chest wall/rib.[36,47,48,49]
- Head and neck.[50]
- Spine/sacrum.[51,52,53,54]
High-Dose Chemotherapy With Stem Cell Support for Ewing Sarcoma
For patients with a high risk of relapse with conventional treatments, some investigators have used high-dose chemotherapy with hematopoietic stem cell transplant (HSCT) as consolidation treatment, in an effort to improve outcome.[19,55,56,57,58,59,60,61,62,63,64,65,66,67]
Evidence (high-dose therapy with stem cell support):
- In a prospective study, patients with bone and/or bone marrow metastases at diagnosis were treated with aggressive chemotherapy, surgery, and/or radiation therapy and HSCT if a good initial response was achieved.[60]
- The study showed no benefit for HSCT compared with historical controls.
- A retrospective review using international bone marrow transplant registries compared the outcomes after treatment with either reduced-intensity conditioning or high-intensity conditioning followed by allogeneic HSCT for patients with Ewing sarcoma at high risk of relapse.[68][Level of evidence C1]
- There was no difference in outcome, and the authors concluded that this suggested the absence of a clinically relevant graft-versus-tumor effect against Ewing sarcoma tumor cells with current approaches.
- The role of high-dose therapy with busulfan-melphalan (BuMel) followed by stem cell rescue was investigated in the prospective randomized EURO-EWING-INTERGROUP-EE99 (NCT00020566) trial for two distinct groups:[69]
- Patients who presented with isolated pulmonary metastases (R2pulm).
- Patients with localized tumors with poor response to initial chemotherapy (<90% necrosis) or with large tumors (>200 mL) (R2loc).
Both study arms were compromised by the potential for selection bias for patients who were eligible for and accepted randomization, which may limit the generalizability of the results. Only 40% of eligible patients were randomized.
- For R2pulm patients, there was no statistically significant difference in EFS or OS between the treatment groups.[70]
- The EFS rates at 3 years were 50.6% for patients who received VAI plus whole-lung irradiation versus 56.6% for patients who received BuMel. The EFS rates at 8 years were 43.1% for patients who received VAI plus whole-lung irradiation versus 52.9% for patients who received BuMel.
- The OS rates at 3 years were 68.0% for patients who received VAI plus whole-lung irradiation versus 68.2% for patients who received BuMel. The OS rates at 8 years were 54.2% for patients who received VAI plus whole-lung irradiation versus 55.3% for patients who received BuMel.
- Among R2loc patients, the 3-year EFS rate was superior with BuMel compared with continued chemotherapy (66.9% vs. 53.1%; P = .019). The 3-year OS rate was 78.0% with BuMel and 72.2% with continued chemotherapy (P = .028).[69]
The induction regimen employed in the EURO-EWING-INTERGROUP-EE99 trial was VIDE. This regimen is less dose intensive than the regimen employed in COG studies. This can be inferred from the intended dose intensity of the agents employed for the 21-week period that preceded randomization in the EURO-EWING-INTERGROUP-EE99 study (see Table 6). The lower dose intensity can also be inferred from the outcome of the EURO-EWING-INTERGROUP-EE99 study for patients in the localized disease stratum. Results from this study include the following:
- Patients assigned to the most favorable risk stratum, R1, were patients with small primary tumors, less than 200 mL in volume. In addition, patients who had poor response to the initial six cycles of therapy with VIDE, as assessed by pathology or radiology, were removed from the R1 stratum and assigned to the R2 stratum. As a result, the R1 stratum includes only patients with small primary tumors and favorable response to initial therapy. The probability for EFS at 3 years for this favorable group was 76%, and the OS rate at 3 years was 85%.[28]
- For all patients with localized Ewing sarcoma, including patients with large primary tumors and patients with poor response to initial therapy treated on the COG-AEWS1031 (NCT01231906) trial, the 5-year probability for EFS was 73%, and the 5-year OS rate was 88%.[24]
The observation that high-dose therapy with autologous stem cell rescue improved outcomes for patients with a poor response to initial therapy in the EURO-EWING-INTERGROUP-EE99 study must be interpreted in this context. The advantage of high-dose therapy as consolidation for patients with a poor response to initial treatment with a less intensive regimen cannot be extrapolated to a population of patients who received a more intensive treatment regimen as initial therapy.
Table 6. Comparison of the Dose Intensity of the EURO-EWING-INTERGROUP-EE99 Trial Versus COG Interval Dose Compression| Chemotherapy Agent | Prescribed Dose Intensity (mg/week) |
|---|
| | EURO-EWING-INTERGROUP-EE99 Trial[28] | COG Interval Dose Compression[24] |
|---|
| COG = Children's Oncology Group. |
| Vincristine | 0.5 mg/m2 | 0.43 mg/m2 |
| Doxorubicin | 17.1 mg/m2 | 21.4 mg/m2 |
| Ifosfamide | 3,000 mg/m2 | 2,150 mg/m2 |
| Cyclophosphamide | 0 | 343 mg/m2 |
| Cyclophosphamide equivalent dose (= cyclophosphamide dose + ifosfamide dose × 0.244) | 732 mg/m2 | 868 mg/m2 |
- Multiple small studies that report benefit for HSCT have been published but are difficult to interpret because only patients who have a good initial response to standard chemotherapy are considered for HSCT.
Extraosseous Ewing Sarcoma
Extraosseous Ewing sarcoma is biologically similar to Ewing sarcoma arising in bone. Historically, most children and young adults with extraosseous Ewing sarcoma were treated on protocols designed for the treatment of rhabdomyosarcoma. This is important because many of the treatment regimens for rhabdomyosarcoma do not include an anthracycline, which is a critical component of current treatment regimens for Ewing sarcoma. Currently, patients with extraosseous Ewing sarcoma are eligible for studies that include Ewing sarcoma of bone.
Evidence (treatment of extraosseous Ewing sarcoma):
- From 1987 to 2004, 111 patients with nonmetastatic extraosseous Ewing sarcoma were enrolled on the RMS-88 and RMS-96 protocols.[71] Patients with initial complete tumor resection received ifosfamide, vincristine, and actinomycin (IVA) while patients with residual tumor received IVA plus doxorubicin (VAIA) or IVA plus carboplatin, epirubicin, and etoposide (CEVAIE). Seventy-six percent of patients received radiation.
- The 5-year EFS rate was 59%, and the OS rate was 69%.
- In a multivariate analysis, independent adverse prognostic factors included axial primary, tumor size greater than 10 cm, Intergroup Rhabdomyosarcoma Studies Group III, and lack of radiation therapy.
- In a retrospective French study, patients with extraosseous Ewing sarcoma were treated using a rhabdomyosarcoma regimen (no anthracyclines) or a Ewing sarcoma regimen (includes anthracyclines).[72,73]
- Patients who received the anthracycline-containing regimen had a significantly better EFS and OS than patients who did not receive anthracyclines.
- Two North American Ewing sarcoma trials included patients with extraosseous Ewing sarcoma.[24,74] In a review of data from the POG-9354 (INT-0154) and EWS0031 (NCT00006734) studies, 213 patients with extraosseous Ewing sarcoma and 826 patients with Ewing sarcoma of bone were identified.[75][Level of evidence C2]
- The HR for EFS of extraosseous Ewing sarcoma was superior (0.62), and extraosseous Ewing sarcoma was a favorable risk factor, independent of age, race, and primary site.
- In addition to the above review, the COG AEWS1031 (NCT01231906) trial subsequently treated patients using a compressed chemotherapy schedule (every 2 weeks). Patients were randomly assigned to receive standard cycles of vincristine, doxorubicin, and cyclophosphamide, alternating with either ifosfamide and etoposide or vincristine, topotecan and cyclophosphamide every third cycle. There were 116 patients with extraosseous tumors, 114 patients with pelvic bone tumors, and 399 patients with nonpelvic bone tumors.[26]
- There were no differences in patient outcomes between the treatment regimens.
- The 5-year EFS rates were 75% (95% CI, 65%–82%) for patients with pelvic bone tumors, 78% (95% CI, 73%–81%) for patients with nonpelvic bone tumors, and 85% (95% CI, 76%–90%) for patients with extraosseous primary sites (global P value = .124).
- The Cooperative Weichteilsarkomstudiengruppe (CWS) performed a retrospective analysis of 243 patients with nonmetastatic extraosseous Ewing sarcoma treated on three consecutive soft tissue sarcoma studies between 1991 and 2008. Several different drug regimens were used, all containing vincristine, doxorubicin, and alkylating agents.[76]
- The outcome improved over time, but it was difficult to assign causality to any specific therapy with these historical comparisons.
- Patients with extremity primary tumors (compared with other locations) and patients with smaller tumors had better outcomes.
- The 5-year EFS rate varied, from 57% to 79%, with the better outcome seen in the most recent protocol.
Cutaneous Ewing sarcoma is a soft tissue tumor in the skin or subcutaneous tissue that seems to behave as a less-aggressive tumor than primary bone or soft tissue Ewing sarcoma. Tumors can form throughout the body, although the extremity is the most common site, and they are almost always localized.
Evidence (treatment of cutaneous Ewing sarcoma):
- In a review of 78 reported cases (some lacking molecular confirmation), the OS rate was 91%. Adequate local control, defined as a complete resection with negative margins, radiation therapy, or a combination, significantly reduced the incidence of relapse. Standard chemotherapy for Ewing sarcoma is often used for these patients because there are no data to suggest which patients could be treated less aggressively.[77,78]
- A series of 56 patients with cutaneous or subcutaneous Ewing sarcoma confirmed the excellent outcome with the use of standard systemic therapy and local control. Attempted primary definitive surgery often resulted in the need for either radiation therapy or more function-compromising surgery, supporting the recommendation of biopsy only as initial surgery, rather than up-front unplanned resection.[79][Level of evidence C2]
References:
- Fuchs B, Valenzuela RG, Sim FH: Pathologic fracture as a complication in the treatment of Ewing's sarcoma. Clin Orthop (415): 25-30, 2003.
- Singh VM, Salunga RC, Huang VJ, et al.: Analysis of the effect of various decalcification agents on the quantity and quality of nucleic acid (DNA and RNA) recovered from bone biopsies. Ann Diagn Pathol 17 (4): 322-6, 2013.
- Hoffer FA, Gianturco LE, Fletcher JA, et al.: Percutaneous biopsy of peripheral primitive neuroectodermal tumors and Ewing's sarcomas for cytogenetic analysis. AJR Am J Roentgenol 162 (5): 1141-2, 1994.
- Craft A, Cotterill S, Malcolm A, et al.: Ifosfamide-containing chemotherapy in Ewing's sarcoma: The Second United Kingdom Children's Cancer Study Group and the Medical Research Council Ewing's Tumor Study. J Clin Oncol 16 (11): 3628-33, 1998.
- Shankar AG, Pinkerton CR, Atra A, et al.: Local therapy and other factors influencing site of relapse in patients with localised Ewing's sarcoma. United Kingdom Children's Cancer Study Group (UKCCSG). Eur J Cancer 35 (12): 1698-704, 1999.
- Nilbert M, Saeter G, Elomaa I, et al.: Ewing's sarcoma treatment in Scandinavia 1984-1990--ten-year results of the Scandinavian Sarcoma Group Protocol SSGIV. Acta Oncol 37 (4): 375-8, 1998.
- Ferrari S, Mercuri M, Rosito P, et al.: Ifosfamide and actinomycin-D, added in the induction phase to vincristine, cyclophosphamide and doxorubicin, improve histologic response and prognosis in patients with non metastatic Ewing's sarcoma of the extremity. J Chemother 10 (6): 484-91, 1998.
- Grier HE, Krailo MD, Tarbell NJ, et al.: Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 348 (8): 694-701, 2003.
- Thacker MM, Temple HT, Scully SP: Current treatment for Ewing's sarcoma. Expert Rev Anticancer Ther 5 (2): 319-31, 2005.
- Juergens C, Weston C, Lewis I, et al.: Safety assessment of intensive induction with vincristine, ifosfamide, doxorubicin, and etoposide (VIDE) in the treatment of Ewing tumors in the EURO-E.W.I.N.G. 99 clinical trial. Pediatr Blood Cancer 47 (1): 22-9, 2006.
- Dunst J, Schuck A: Role of radiotherapy in Ewing tumors. Pediatr Blood Cancer 42 (5): 465-70, 2004.
- Donaldson SS: Ewing sarcoma: radiation dose and target volume. Pediatr Blood Cancer 42 (5): 471-6, 2004.
- Bacci G, Ferrari S, Longhi A, et al.: Role of surgery in local treatment of Ewing's sarcoma of the extremities in patients undergoing adjuvant and neoadjuvant chemotherapy. Oncol Rep 11 (1): 111-20, 2004.
- Krasin MJ, Rodriguez-Galindo C, Davidoff AM, et al.: Efficacy of combined surgery and irradiation for localized Ewings sarcoma family of tumors. Pediatr Blood Cancer 43 (3): 229-36, 2004.
- Bacci G, Longhi A, Briccoli A, et al.: The role of surgical margins in treatment of Ewing's sarcoma family tumors: experience of a single institution with 512 patients treated with adjuvant and neoadjuvant chemotherapy. Int J Radiat Oncol Biol Phys 65 (3): 766-72, 2006.
- Paulussen M, Ahrens S, Burdach S, et al.: Primary metastatic (stage IV) Ewing tumor: survival analysis of 171 patients from the EICESS studies. European Intergroup Cooperative Ewing Sarcoma Studies. Ann Oncol 9 (3): 275-81, 1998.
- Pinkerton CR, Bataillard A, Guillo S, et al.: Treatment strategies for metastatic Ewing's sarcoma. Eur J Cancer 37 (11): 1338-44, 2001.
- Bernstein ML, Devidas M, Lafreniere D, et al.: Intensive therapy with growth factor support for patients with Ewing tumor metastatic at diagnosis: Pediatric Oncology Group/Children's Cancer Group Phase II Study 9457--a report from the Children's Oncology Group. J Clin Oncol 24 (1): 152-9, 2006.
- Ladenstein R, Pötschger U, Le Deley MC, et al.: Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol 28 (20): 3284-91, 2010.
- Miser JS, Krailo MD, Tarbell NJ, et al.: Treatment of metastatic Ewing's sarcoma or primitive neuroectodermal tumor of bone: evaluation of combination ifosfamide and etoposide--a Children's Cancer Group and Pediatric Oncology Group study. J Clin Oncol 22 (14): 2873-6, 2004.
- Miser JS, Goldsby RE, Chen Z, et al.: Treatment of metastatic Ewing sarcoma/primitive neuroectodermal tumor of bone: evaluation of increasing the dose intensity of chemotherapy--a report from the Children's Oncology Group. Pediatr Blood Cancer 49 (7): 894-900, 2007.
- Haeusler J, Ranft A, Boelling T, et al.: The value of local treatment in patients with primary, disseminated, multifocal Ewing sarcoma (PDMES). Cancer 116 (2): 443-50, 2010.
- Paulussen M, Craft AW, Lewis I, et al.: Results of the EICESS-92 Study: two randomized trials of Ewing's sarcoma treatment--cyclophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in high-risk patients. J Clin Oncol 26 (27): 4385-93, 2008.
- Womer RB, West DC, Krailo MD, et al.: Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 30 (33): 4148-54, 2012.
- Cash T, Krailo MD, Buxton AB, et al.: Long-Term Outcomes in Patients With Localized Ewing Sarcoma Treated With Interval-Compressed Chemotherapy on Children's Oncology Group Study AEWS0031. J Clin Oncol 41 (30): 4724-4728, 2023.
- Leavey PJ, Laack NN, Krailo MD, et al.: Phase III Trial Adding Vincristine-Topotecan-Cyclophosphamide to the Initial Treatment of Patients With Nonmetastatic Ewing Sarcoma: A Children's Oncology Group Report. J Clin Oncol 39 (36): 4029-4038, 2021.
- Brennan B, Kirton L, Marec-Bérard P, et al.: Comparison of two chemotherapy regimens in patients with newly diagnosed Ewing sarcoma (EE2012): an open-label, randomised, phase 3 trial. Lancet 400 (10362): 1513-1521, 2022.
- Le Deley MC, Paulussen M, Lewis I, et al.: Cyclophosphamide compared with ifosfamide in consolidation treatment of standard-risk Ewing sarcoma: results of the randomized noninferiority Euro-EWING99-R1 trial. J Clin Oncol 32 (23): 2440-8, 2014.
- Brunetto AL, Castillo LA, Petrilli AS, et al.: Carboplatin in the treatment of Ewing sarcoma: Results of the first Brazilian collaborative study group for Ewing sarcoma family tumors-EWING1. Pediatr Blood Cancer 62 (10): 1747-53, 2015.
- DuBois SG, Krailo MD, Gebhardt MC, et al.: Comparative evaluation of local control strategies in localized Ewing sarcoma of bone: a report from the Children's Oncology Group. Cancer 121 (3): 467-75, 2015.
- Lin TA, Ludmir EB, Liao KP, et al.: Timing of Local Therapy Affects Survival in Ewing Sarcoma. Int J Radiat Oncol Biol Phys 104 (1): 127-136, 2019.
- Yock TI, Krailo M, Fryer CJ, et al.: Local control in pelvic Ewing sarcoma: analysis from INT-0091--a report from the Children's Oncology Group. J Clin Oncol 24 (24): 3838-43, 2006.
- Andreou D, Ranft A, Gosheger G, et al.: Which Factors Are Associated with Local Control and Survival of Patients with Localized Pelvic Ewing's Sarcoma? A Retrospective Analysis of Data from the Euro-EWING99 Trial. Clin Orthop Relat Res 478 (2): 290-302, 2020.
- Foulon S, Brennan B, Gaspar N, et al.: Can postoperative radiotherapy be omitted in localised standard-risk Ewing sarcoma? An observational study of the Euro-E.W.I.N.G group. Eur J Cancer 61: 128-36, 2016.
- Seitz G, Urla C, Sparber-Sauer M, et al.: Treatment and outcome of patients with thoracic tumors of the Ewing sarcoma family: A report from the Cooperative Weichteilsarkom Studiengruppe CWS-81, -86, -91, -96, and -2002P trials. Pediatr Blood Cancer 66 (3): e27537, 2019.
- Shamberger RC, LaQuaglia MP, Gebhardt MC, et al.: Ewing sarcoma/primitive neuroectodermal tumor of the chest wall: impact of initial versus delayed resection on tumor margins, survival, and use of radiation therapy. Ann Surg 238 (4): 563-7; discussion 567-8, 2003.
- Wagner TD, Kobayashi W, Dean S, et al.: Combination short-course preoperative irradiation, surgical resection, and reduced-field high-dose postoperative irradiation in the treatment of tumors involving the bone. Int J Radiat Oncol Biol Phys 73 (1): 259-66, 2009.
- Hoffmann C, Ahrens S, Dunst J, et al.: Pelvic Ewing sarcoma: a retrospective analysis of 241 cases. Cancer 85 (4): 869-77, 1999.
- Sucato DJ, Rougraff B, McGrath BE, et al.: Ewing's sarcoma of the pelvis. Long-term survival and functional outcome. Clin Orthop (373): 193-201, 2000.
- Bacci G, Ferrari S, Mercuri M, et al.: Multimodal therapy for the treatment of nonmetastatic Ewing sarcoma of pelvis. J Pediatr Hematol Oncol 25 (2): 118-24, 2003.
- Bacci G, Ferrari S, Longhi A, et al.: Local and systemic control in Ewing's sarcoma of the femur treated with chemotherapy, and locally by radiotherapy and/or surgery. J Bone Joint Surg Br 85 (1): 107-14, 2003.
- Ozaki T, Hillmann A, Hoffmann C, et al.: Ewing's sarcoma of the femur. Prognosis in 69 patients treated by the CESS group. Acta Orthop Scand 68 (1): 20-4, 1997.
- Ayoub KS, Fiorenza F, Grimer RJ, et al.: Extensible endoprostheses of the humerus after resection of bone tumours. J Bone Joint Surg Br 81 (3): 495-500, 1999.
- Bacci G, Palmerini E, Staals EL, et al.: Ewing's sarcoma family tumors of the humerus: outcome of patients treated with radiotherapy, surgery or surgery and adjuvant radiotherapy. Radiother Oncol 93 (2): 383-7, 2009.
- Casadei R, Magnani M, Biagini R, et al.: Prognostic factors in Ewing's sarcoma of the foot. Clin Orthop (420): 230-8, 2004.
- Anakwenze OA, Parker WL, Wold LE, et al.: Ewing's sarcoma of the hand. J Hand Surg Eur Vol 34 (1): 35-9, 2009.
- Shamberger RC, Laquaglia MP, Krailo MD, et al.: Ewing sarcoma of the rib: results of an intergroup study with analysis of outcome by timing of resection. J Thorac Cardiovasc Surg 119 (6): 1154-61, 2000.
- Sirvent N, Kanold J, Levy C, et al.: Non-metastatic Ewing's sarcoma of the ribs: the French Society of Pediatric Oncology Experience. Eur J Cancer 38 (4): 561-7, 2002.
- Schuck A, Ahrens S, Konarzewska A, et al.: Hemithorax irradiation for Ewing tumors of the chest wall. Int J Radiat Oncol Biol Phys 54 (3): 830-8, 2002.
- Windfuhr JP: Primitive neuroectodermal tumor of the head and neck: incidence, diagnosis, and management. Ann Otol Rhinol Laryngol 113 (7): 533-43, 2004.
- Venkateswaran L, Rodriguez-Galindo C, Merchant TE, et al.: Primary Ewing tumor of the vertebrae: clinical characteristics, prognostic factors, and outcome. Med Pediatr Oncol 37 (1): 30-5, 2001.
- Marco RA, Gentry JB, Rhines LD, et al.: Ewing's sarcoma of the mobile spine. Spine 30 (7): 769-73, 2005.
- Schuck A, Ahrens S, von Schorlemer I, et al.: Radiotherapy in Ewing tumors of the vertebrae: treatment results and local relapse analysis of the CESS 81/86 and EICESS 92 trials. Int J Radiat Oncol Biol Phys 63 (5): 1562-7, 2005.
- Bacci G, Boriani S, Balladelli A, et al.: Treatment of nonmetastatic Ewing's sarcoma family tumors of the spine and sacrum: the experience from a single institution. Eur Spine J 18 (8): 1091-5, 2009.
- Kushner BH, Meyers PA: How effective is dose-intensive/myeloablative therapy against Ewing's sarcoma/primitive neuroectodermal tumor metastatic to bone or bone marrow? The Memorial Sloan-Kettering experience and a literature review. J Clin Oncol 19 (3): 870-80, 2001.
- Marina N, Meyers PA: High-dose therapy and stem-cell rescue for Ewing's family of tumors in second remission. J Clin Oncol 23 (19): 4262-4, 2005.
- Burdach S: Treatment of advanced Ewing tumors by combined radiochemotherapy and engineered cellular transplants. Pediatr Transplant 8 (Suppl 5): 67-82, 2004.
- McTiernan A, Driver D, Michelagnoli MP, et al.: High dose chemotherapy with bone marrow or peripheral stem cell rescue is an effective treatment option for patients with relapsed or progressive Ewing's sarcoma family of tumours. Ann Oncol 17 (8): 1301-5, 2006.
- Burdach S, Meyer-Bahlburg A, Laws HJ, et al.: High-dose therapy for patients with primary multifocal and early relapsed Ewing's tumors: results of two consecutive regimens assessing the role of total-body irradiation. J Clin Oncol 21 (16): 3072-8, 2003.
- Meyers PA, Krailo MD, Ladanyi M, et al.: High-dose melphalan, etoposide, total-body irradiation, and autologous stem-cell reconstitution as consolidation therapy for high-risk Ewing's sarcoma does not improve prognosis. J Clin Oncol 19 (11): 2812-20, 2001.
- Oberlin O, Rey A, Desfachelles AS, et al.: Impact of high-dose busulfan plus melphalan as consolidation in metastatic Ewing tumors: a study by the Société Française des Cancers de l'Enfant. J Clin Oncol 24 (24): 3997-4002, 2006.
- Hawkins D, Barnett T, Bensinger W, et al.: Busulfan, melphalan, and thiotepa with or without total marrow irradiation with hematopoietic stem cell rescue for poor-risk Ewing-Sarcoma-Family tumors. Med Pediatr Oncol 34 (5): 328-37, 2000.
- Rosenthal J, Bolotin E, Shakhnovits M, et al.: High-dose therapy with hematopoietic stem cell rescue in patients with poor prognosis Ewing family tumors. Bone Marrow Transplant 42 (5): 311-8, 2008.
- Burdach S, Thiel U, Schöniger M, et al.: Total body MRI-governed involved compartment irradiation combined with high-dose chemotherapy and stem cell rescue improves long-term survival in Ewing tumor patients with multiple primary bone metastases. Bone Marrow Transplant 45 (3): 483-9, 2010.
- Gaspar N, Rey A, Bérard PM, et al.: Risk adapted chemotherapy for localised Ewing's sarcoma of bone: the French EW93 study. Eur J Cancer 48 (9): 1376-85, 2012.
- Drabko K, Raciborska A, Bilska K, et al.: Consolidation of first-line therapy with busulphan and melphalan, and autologous stem cell rescue in children with Ewing's sarcoma. Bone Marrow Transplant 47 (12): 1530-4, 2012.
- Loschi S, Dufour C, Oberlin O, et al.: Tandem high-dose chemotherapy strategy as first-line treatment of primary disseminated multifocal Ewing sarcomas in children, adolescents and young adults. Bone Marrow Transplant 50 (8): 1083-8, 2015.
- Thiel U, Wawer A, Wolf P, et al.: No improvement of survival with reduced- versus high-intensity conditioning for allogeneic stem cell transplants in Ewing tumor patients. Ann Oncol 22 (7): 1614-21, 2011.
- Whelan J, Le Deley MC, Dirksen U, et al.: High-Dose Chemotherapy and Blood Autologous Stem-Cell Rescue Compared With Standard Chemotherapy in Localized High-Risk Ewing Sarcoma: Results of Euro-E.W.I.N.G.99 and Ewing-2008. J Clin Oncol : JCO2018782516, 2018.
- Dirksen U, Brennan B, Le Deley MC, et al.: High-Dose Chemotherapy Compared With Standard Chemotherapy and Lung Radiation in Ewing Sarcoma With Pulmonary Metastases: Results of the European Ewing Tumour Working Initiative of National Groups, 99 Trial and EWING 2008. J Clin Oncol 37 (34): 3192-3202, 2019.
- Spiller M, Bisogno G, Ferrari A, et al.: Prognostic factors in localized extraosseus Ewing family tumors. [Abstract] Pediatr Blood Cancer 46 (10) : A-PD.024, 434, 2006.
- Castex MP, Rubie H, Stevens MC, et al.: Extraosseous localized ewing tumors: improved outcome with anthracyclines--the French society of pediatric oncology and international society of pediatric oncology. J Clin Oncol 25 (10): 1176-82, 2007.
- Dantonello TM, Int-Veen C, Harms D, et al.: Cooperative trial CWS-91 for localized soft tissue sarcoma in children, adolescents, and young adults. J Clin Oncol 27 (9): 1446-55, 2009.
- Granowetter L, Womer R, Devidas M, et al.: Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: a Children's Oncology Group Study. J Clin Oncol 27 (15): 2536-41, 2009.
- Cash T, McIlvaine E, Krailo MD, et al.: Comparison of clinical features and outcomes in patients with extraskeletal versus skeletal localized Ewing sarcoma: A report from the Children's Oncology Group. Pediatr Blood Cancer 63 (10): 1771-9, 2016.
- Koscielniak E, Sparber-Sauer M, Scheer M, et al.: Extraskeletal Ewing sarcoma in children, adolescents, and young adults. An analysis of three prospective studies of the Cooperative Weichteilsarkomstudiengruppe (CWS). Pediatr Blood Cancer 68 (10): e29145, 2021.
- Collier AB, Simpson L, Monteleone P: Cutaneous Ewing sarcoma: report of 2 cases and literature review of presentation, treatment, and outcome of 76 other reported cases. J Pediatr Hematol Oncol 33 (8): 631-4, 2011.
- Terrier-Lacombe MJ, Guillou L, Chibon F, et al.: Superficial primitive Ewing's sarcoma: a clinicopathologic and molecular cytogenetic analysis of 14 cases. Mod Pathol 22 (1): 87-94, 2009.
- Di Giannatale A, Frezza AM, Le Deley MC, et al.: Primary cutaneous and subcutaneous Ewing sarcoma. Pediatr Blood Cancer 62 (9): 1555-61, 2015.
Special Considerations for the Treatment of Children With Cancer
Cancer in children and adolescents is rare, although the overall incidence has slowly increased since 1975.[1] Children and adolescents with cancer should be referred to medical centers that have a multidisciplinary team of cancer specialists with experience treating the cancers that occur during childhood and adolescence. This multidisciplinary team approach incorporates the skills of the following pediatric specialists and others to ensure that children receive treatment, supportive care, and rehabilitation to achieve optimal survival and quality of life:
- Primary care physicians.
- Pediatric surgeons.
- Transplant surgeons.
- Pathologists.
- Pediatric radiation oncologists.
- Pediatric medical oncologists and hematologists.
- Ophthalmologists.
- Rehabilitation specialists.
- Pediatric oncology nurses.
- Social workers.
- Child-life professionals.
- Psychologists.
- Nutritionists.
For specific information about supportive care for children and adolescents with cancer, see the summaries on Supportive and Palliative Care.
The American Academy of Pediatrics has outlined guidelines for pediatric cancer centers and their role in the treatment of children and adolescents with cancer.[2] At these centers, clinical trials are available for most types of cancer that occur in children and adolescents, and the opportunity to participate is offered to most patients and their families. Clinical trials for children and adolescents diagnosed with cancer are generally designed to compare potentially better therapy with current standard therapy. Other types of clinical trials test novel therapies when there is no standard therapy for a cancer diagnosis. Most of the progress in identifying curative therapies for childhood cancers has been achieved through clinical trials. Information about ongoing clinical trials is available from the NCI website.
Childhood and adolescent cancer survivors require close monitoring because side effects of cancer therapy may persist or develop months or years after treatment. For specific information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors, see Late Effects of Treatment for Childhood Cancer.
References:
- Smith MA, Seibel NL, Altekruse SF, et al.: Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol 28 (15): 2625-34, 2010.
- American Academy of Pediatrics: Standards for pediatric cancer centers. Pediatrics 134 (2): 410-4, 2014. Also available online. Last accessed August 23, 2024.
Treatment of Localized Ewing Sarcoma
Standard treatment options for localized Ewing sarcoma include the following:
- Chemotherapy.
- Local-control measures:
- High-dose chemotherapy with autologous stem cell rescue.
Because most patients with apparently localized disease at diagnosis have occult metastatic disease, multidrug chemotherapy and surgery and/or radiation therapy (to control local disease) is indicated in the treatment of all patients.[1,2,3,4,5,6,7,8] Patients with localized Ewing sarcoma who receive current treatment regimens achieve event-free survival (EFS) and overall survival (OS) rates of approximately 70% at 5 years after diagnosis.[9]
Chemotherapy
Current standard chemotherapy in the United States includes vincristine, doxorubicin, and cyclophosphamide (VDC), alternating with ifosfamide and etoposide (IE) or VDC/IE.[9]; [10][Level of evidence A1] With the outcome of the EURO EWING 2012 (EE2012) trial, which compared VDC/IE to VIDE (vincristine, ifosfamide, doxorubicin, etoposide), the VDC/IE regimen has become increasingly used internationally as initial therapy over the previous VIDE regimen.[11][Level of evidence B1] For more information about the EE2012 trial, see the Chemotherapy for Ewing Sarcoma section.
Evidence (chemotherapy):
- IE has shown activity in Ewing sarcoma. A large randomized clinical trial and a nonrandomized trial demonstrated that outcome was improved when IE was alternated with VDC.[9,12]
- The use of high-dose VDC has shown promising results in small numbers of patients. A single-institution study of 44 patients treated with high-dose VDC and IE showed a 4-year EFS rate of 82%.[13]
- However, in an intergroup trial of the Pediatric Oncology Group and the Children's Cancer Group, which compared an alkylator dose-intensified VDC/IE regimen with standard alkylator doses of the same VDC/IE regimen, no differences in outcome were observed.[14] Unlike the single-institution trial, this trial did not maintain the dose intensity of cyclophosphamide for the duration of treatment.[13]
- In a Children's Oncology Group (COG) trial (COG-AEWS0031 [NCT00006734]), 568 patients with newly diagnosed localized extradural Ewing sarcoma were randomly assigned to receive chemotherapy (VDC/IE) given either every 2 weeks (interval compression) or every 3 weeks (standard).[10]
- Patients randomly assigned to the every-2-week interval of treatment had an improved 5-year EFS rate (73% vs. 65%, P = .048).
- There was no increase in toxicity observed with the every-2-week schedule.
- With longer follow-up of 10 years, interval-compressed VDC/IE continued to demonstrate superior EFS. OS was also significantly higher with this regimen than with VDC/IE given every 3 weeks.[15]
- The European Ewing 2008R1 trial evaluated 284 patients with standard-risk Ewing sarcoma, defined as localized disease with favorable histological response to initial chemotherapy and/or an initial tumor volume of less than 200 mL. All patients received VIDE chemotherapy followed by vincristine, dactinomycin, and ifosfamide (VAI) (male) or vincristine, dactinomycin, and cyclophosphamide (VAC) (female) consolidation therapy. During the sixth cycle of consolidation, patients were randomly assigned to therapy with either the addition of zoledronic acid for nine 28-day cycles or no maintenance therapy.[16]
- Outcomes were similar between patients who received zoledronic acid and patients who did not receive maintenance therapy (3-year EFS rates, 84.0% vs. 81.7%).
- Patients who received zoledronic acid experienced higher rates of renal, neurological, and gastrointestinal toxicities.
- EE2012 was a European investigator–initiated, open-label, randomized, controlled, phase III trial that took place in ten countries. Between 2014 and 2019, 640 patients were enrolled and were randomly allocated (1:1) to either the European or COG treatment regimens (320 patients in each treatment group). The European treatment regimen for induction included VIDE, and the consolidation regimen included vincristine, dactinomycin, with ifosfamide or cyclophosphamide, or busulfan and melphalan (group 1). The COG treatment regimen for induction included interval-compressed VDC/IE, and the consolidation regimen included vincristine and cyclophosphamide, with ifosfamide and etoposide or busulfan and melphalan (group 2).[11][Level of evidence B1]
- The 3-year EFS rate was 61% for patients in group 1 and 67% for patients in group 2 (adjusted hazard ratio [HR], 0.71; 95% confidence interval [CI], 0.55–0.92 in favor of group 2).
- The probability that the true HR for group 2 with interval-compressed VDC/IE was less than 1 was greater than 0.99.
- The investigators concluded that dose-intensive induction chemotherapy with the VDC/IE regimen was more effective, less toxic, and shorter in duration for patients with all stages of newly diagnosed Ewing sarcoma than the VIDE regimen.
Local-Control Measures
Local control can be achieved by surgery and/or radiation therapy. Decisions regarding the optimal modality for local control for an individual patient involve consideration of the following:
- The possibility of complete resection with adequate margins after an initial period of systemic therapy.
- The predicted functional impact of a surgical procedure.
- The predicted morbidity after radiation therapy.
- The possibility of increased risk of second malignant neoplasms after radiation therapy.
An analysis using propensity scoring (a method that adjusts for the inherent selection bias of the location and size of the tumor) to adjust for clinical features that may influence the preference for surgery only, radiation only, or combined surgery and radiation demonstrated that similar EFS rates are achieved with each mode of local therapy after propensity adjustment.[17]
Surgery
Surgery is generally the preferred approach if the lesion is resectable.[18,19] The superiority of resection for local control has never been tested in a prospective randomized trial. The apparent superiority may represent selection bias.
- In past studies, smaller, more peripheral tumors were more likely to be treated with surgery, and larger, more central tumors were more likely to be treated with radiation therapy.[20]
- An Italian retrospective study showed that surgery improved outcome only in extremity tumors, although the number of patients with central axis Ewing sarcoma who achieved adequate margins was small.[8]
- In a series of 39 patients who received both surgery and radiation therapy at St. Jude Children's Research Hospital, the 8-year local failure rate was 5% for patients with negative surgical margins and 17% for those with positive margins.[5]
- Data for patients with pelvic primary Ewing sarcoma from a North American intergroup trial showed no difference in local control or survival based on local-control modality—surgery alone, radiation therapy alone, or radiation plus surgery.[21]
- Patients with residual viable tumor in the resected specimen have a worse outcome than those with complete necrosis. In a French Ewing sarcoma study (EW88), the EFS rates were 75% for patients with less than 5% viable tumor, 48% for patients with 5% to 30% viable tumor, and 20% for patients with more than 30% viable tumor.[20]
A single-institution retrospective analysis of 78 patients with Ewing sarcoma suggested that pathological fracture at initial presentation was associated with inferior EFS and OS.[22][Level of evidence C1] Another study found that pathological fracture at the time of diagnosis did not preclude surgical resection and was not associated with adverse outcome.[23]
Radiation therapy
Radiation therapy is usually employed in the following cases:
- Patients who do not have a surgical option that preserves function and cosmesis.
- Patients whose tumors have been excised but with inadequate margins.
- Preoperative radiation therapy if gross-total resection is possible but without adequate margins (and preservation of function and cosmesis).
Radiation therapy is delivered in a setting in which stringent planning techniques are applied by those experienced in the treatment of Ewing sarcoma. Such an approach will result in local control of the tumor with acceptable morbidity in most patients.[1,2,24]
The radiation dose may be adjusted depending on the extent of residual disease after the initial surgical procedure. When no surgical resection is performed, radiation therapy is generally administered in fractionated doses totaling approximately 55.8 Gy to the prechemotherapy tumor volume. A randomized study of 40 patients with Ewing sarcoma using 55.8 Gy to the prechemotherapy tumor extent with a 2-cm margin compared with the same total-tumor dose after 39.6 Gy to the entire bone showed no difference in local control or EFS.[3] Hyperfractionated radiation therapy has not been associated with improved local control or decreased morbidity.[1]
Preoperative radiation therapy is an approach that can be used when surgical resection is deemed possible but with the likelihood of microscopic residual disease. A panel of international expert clinicians used a three-stage modified Delphi technique to develop consensus statements about local treatment. The panel reached a strong consensus that preoperative radiation therapy may be given when an inadequate (marginal) margin at resection is foreseen on imaging.[25]
For patients with residual disease after an attempt at surgical resection, the Intergroup Ewing Sarcoma Study (INT-0091) recommended 45 Gy to the original disease site plus a 10.8 Gy boost for patients with gross residual disease and 45 Gy plus a 5.4 Gy boost for patients with microscopic residual disease. No radiation therapy was recommended for those who had no evidence of microscopic residual disease after surgical resection.[14]
For patients who are deemed to have unresectable disease after induction chemotherapy, radiation therapy is given, using the same doses as those administered for patients with partially resected disease.[26] Patients who have unresectable disease are typically those with extremity tumors that have persistent encasement of the neurovascular bundles and/or morbid surgical excision entailing loss of functionality. In a phase III randomized controlled clinical trial of patients with unresectable disease, patients were randomly assigned to receive either standard-dose radiation therapy (55.8 Gy in 1.8 Gy fractions) or escalated-dose radiation therapy (70.2 Gy in 1.8 Gy fractions).[26] From 2005 to 2015, the study accrued 47 patients who received standard-dose radiation therapy and 48 patients who received escalated-dose radiation therapy (interquartile age, 13–23 years). The median largest tumor dimension was 9.7 cm. At a median follow-up of 67 months, the 5-year local control rate was significantly better in the escalated arm than in the standard arm (76.4% vs. 49.4%; P = .02). The differences in disease-free survival (DFS) and OS at 5 years did not achieve statistical significance (DFS rates, 46.7% vs. 31.8%; P = .22; OS rates, 58.8% vs. 45.4%; P = .08), possibly because the rate of metastatic disease was not changed. A skin toxicity grade of more than 2 was greater in the high-dose arm (10.4% vs. 2.1%; P = .08).
In a single-institution nonrandomized study, patients who had primary tumors 8 cm or larger were treated with higher-dose radiation therapy (median dose, 64.8 Gy). The 5-year cumulative incidence of local failure rate was 6.6%, which compares favorably to other published local failure rates in this group of patients.[27]
Comparison of proton-beam radiation therapy and intensity-modulated radiation therapy (IMRT) treatment plans has shown that proton-beam radiation therapy can spare more normal tissue adjacent to Ewing sarcoma primary tumors than IMRT.[28] Follow-up remains relatively short, and there are no data to determine whether the reduction in dose to adjacent tissue will result in improved functional outcome or reduce the risk of secondary malignancy. Because patient numbers are small and follow-up is relatively short, it is not possible to determine whether the risk of local recurrence might be increased by reducing radiation dose in tissue adjacent to the primary tumor.
Higher rates of local failure are seen in patients older than 14 years who have tumors larger than 8 cm in length.[29] Among patients with pelvic tumors, a larger tumor volume, a periacetabular tumor site, and the use of definitive radiation therapy only (rather than a combined-modality approach) were associated with higher rates of local failure.[30] A retrospective analysis of patients with Ewing sarcoma of the chest wall compared patients who received hemithorax radiation therapy with those who received radiation therapy to the chest wall only. Patients with pleural invasion, pleural effusion, or intraoperative contamination were assigned to hemithorax radiation therapy. EFS was longer for patients who received hemithorax radiation, but the difference was not statistically significant. In addition, most patients with primary vertebral tumors did not receive hemithorax radiation and had a lower probability for EFS.[31]
Radiation therapy is associated with the development of subsequent neoplasms. A retrospective study noted that patients who received 60 Gy or more had an incidence of second malignancy of 20%. Patients who received 48 Gy to 60 Gy had an incidence of 5%, and those who received less than 48 Gy did not develop a second malignancy.[32]
High-Dose Chemotherapy With Autologous Stem Cell Rescue
Evidence (high-dose chemotherapy with autologous stem cell rescue):
- The role of high-dose therapy with busulfan-melphalan (BuMel) followed by stem cell rescue was investigated in the prospective randomized EURO-EWING-INTERGROUP-EE99 (NCT00020566) trial for two distinct groups:[33]
- Patients who presented with isolated pulmonary metastases (R2pulm).
- Patients with localized tumors with poor response to initial chemotherapy (<90% necrosis) or with large tumors (>200 mL) (R2loc).
Both study arms were compromised by the potential for selection bias for patients who were eligible for and accepted randomization, which may limit the generalizability of the results. Only 40% of eligible patients were randomized.
- For R2pulm patients, there was no statistically significant difference in EFS or OS between the treatment groups.[34]
- The EFS rates at 3 years were 50.6% for patients who received vincristine, dactinomycin, and ifosfamide (VAI) plus whole-lung irradiation versus 56.6% for patients who received BuMel.
- The EFS rates at 8 years were 43.1% for patients who received VAI plus whole-lung irradiation versus 52.9% for patients who received BuMel.
- The OS rates at 3 years were 68.0% for patients who received VAI plus whole-lung irradiation versus 68.2% for patients who received BuMel.
- The OS rates at 8 years were 54.2% for patients who received VAI plus whole-lung irradiation versus 55.3% for patients who received BuMel.
- Among R2loc patients, the 3-year EFS rate was superior with BuMel compared with continued chemotherapy (66.9% vs. 53.1%; P = .019). The 3-year OS rate was 78.0% with BuMel and 72.2% with continued chemotherapy (P = .028).[33]
The induction regimen employed in the EURO-EWING-INTERGROUP-EE99 trial included VIDE. This regimen is less dose intensive than the regimen employed in COG studies. This can be inferred from the intended dose intensity of the agents employed for the 21-week period that preceded randomization in the EURO-EWING-INTERGROUP-EE99 study (see Table 6). The lower dose intensity can also be inferred from the outcome of the EURO-EWING-INTERGROUP-EE99 study for patients in the localized disease stratum. Results from this study include the following:
- Patients assigned to the most favorable risk stratum, R1, were patients with small primary tumors, less than 200 mL in volume. In addition, patients who had poor response to the initial six cycles of therapy with VIDE, as assessed by pathology or radiology, were removed from the R1 stratum and assigned to the R2 stratum. As a result, the R1 stratum includes only patients with small primary tumors and favorable response to initial therapy.[35]
- The probability for EFS at 3 years for this favorable group was 76%, and the OS rate at 3 years was 85%.
- For all patients with localized Ewing sarcoma, including patients with large primary tumors and patients with poor response to initial therapy treated on the COG-AEWS1031 (NCT01231906) trial, the 5-year probability for EFS was 73%, and the 5-year OS rate was 88%.[10]
The observation that high-dose therapy with autologous stem cell rescue improved outcomes for patients with a poor response to initial therapy in the EURO-EWING-INTERGROUP-EE99 study must be interpreted in this context. The advantage of high-dose therapy as consolidation for patients with a poor response to initial treatment (with a less intensive regimen) cannot be extrapolated to a population of patients who received a more intensive treatment regimen as initial therapy.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
- Dunst J, Jürgens H, Sauer R, et al.: Radiation therapy in Ewing's sarcoma: an update of the CESS 86 trial. Int J Radiat Oncol Biol Phys 32 (4): 919-30, 1995.
- Donaldson SS, Torrey M, Link MP, et al.: A multidisciplinary study investigating radiotherapy in Ewing's sarcoma: end results of POG #8346. Pediatric Oncology Group. Int J Radiat Oncol Biol Phys 42 (1): 125-35, 1998.
- Craft A, Cotterill S, Malcolm A, et al.: Ifosfamide-containing chemotherapy in Ewing's sarcoma: The Second United Kingdom Children's Cancer Study Group and the Medical Research Council Ewing's Tumor Study. J Clin Oncol 16 (11): 3628-33, 1998.
- Nilbert M, Saeter G, Elomaa I, et al.: Ewing's sarcoma treatment in Scandinavia 1984-1990--ten-year results of the Scandinavian Sarcoma Group Protocol SSGIV. Acta Oncol 37 (4): 375-8, 1998.
- Krasin MJ, Davidoff AM, Rodriguez-Galindo C, et al.: Definitive surgery and multiagent systemic therapy for patients with localized Ewing sarcoma family of tumors: local outcome and prognostic factors. Cancer 104 (2): 367-73, 2005.
- Bacci G, Forni C, Longhi A, et al.: Long-term outcome for patients with non-metastatic Ewing's sarcoma treated with adjuvant and neoadjuvant chemotherapies. 402 patients treated at Rizzoli between 1972 and 1992. Eur J Cancer 40 (1): 73-83, 2004.
- Rosito P, Mancini AF, Rondelli R, et al.: Italian Cooperative Study for the treatment of children and young adults with localized Ewing sarcoma of bone: a preliminary report of 6 years of experience. Cancer 86 (3): 421-8, 1999.
- Bacci G, Longhi A, Briccoli A, et al.: The role of surgical margins in treatment of Ewing's sarcoma family tumors: experience of a single institution with 512 patients treated with adjuvant and neoadjuvant chemotherapy. Int J Radiat Oncol Biol Phys 65 (3): 766-72, 2006.
- Grier HE, Krailo MD, Tarbell NJ, et al.: Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 348 (8): 694-701, 2003.
- Womer RB, West DC, Krailo MD, et al.: Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 30 (33): 4148-54, 2012.
- Brennan B, Kirton L, Marec-Bérard P, et al.: Comparison of two chemotherapy regimens in patients with newly diagnosed Ewing sarcoma (EE2012): an open-label, randomised, phase 3 trial. Lancet 400 (10362): 1513-1521, 2022.
- Ferrari S, Mercuri M, Rosito P, et al.: Ifosfamide and actinomycin-D, added in the induction phase to vincristine, cyclophosphamide and doxorubicin, improve histologic response and prognosis in patients with non metastatic Ewing's sarcoma of the extremity. J Chemother 10 (6): 484-91, 1998.
- Kolb EA, Kushner BH, Gorlick R, et al.: Long-term event-free survival after intensive chemotherapy for Ewing's family of tumors in children and young adults. J Clin Oncol 21 (18): 3423-30, 2003.
- Granowetter L, Womer R, Devidas M, et al.: Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: a Children's Oncology Group Study. J Clin Oncol 27 (15): 2536-41, 2009.
- Cash T, Krailo MD, Buxton AB, et al.: Long-Term Outcomes in Patients With Localized Ewing Sarcoma Treated With Interval-Compressed Chemotherapy on Children's Oncology Group Study AEWS0031. J Clin Oncol 41 (30): 4724-4728, 2023.
- Koch R, Haveman L, Ladenstein R, et al.: Zoledronic Acid Add-on Therapy for Standard-Risk Ewing Sarcoma Patients in the Ewing 2008R1 Trial. Clin Cancer Res 29 (24): 5057-5068, 2023.
- DuBois SG, Krailo MD, Gebhardt MC, et al.: Comparative evaluation of local control strategies in localized Ewing sarcoma of bone: a report from the Children's Oncology Group. Cancer 121 (3): 467-75, 2015.
- Hoffmann C, Ahrens S, Dunst J, et al.: Pelvic Ewing sarcoma: a retrospective analysis of 241 cases. Cancer 85 (4): 869-77, 1999.
- Shamberger RC, Laquaglia MP, Krailo MD, et al.: Ewing sarcoma of the rib: results of an intergroup study with analysis of outcome by timing of resection. J Thorac Cardiovasc Surg 119 (6): 1154-61, 2000.
- Oberlin O, Deley MC, Bui BN, et al.: Prognostic factors in localized Ewing's tumours and peripheral neuroectodermal tumours: the third study of the French Society of Paediatric Oncology (EW88 study). Br J Cancer 85 (11): 1646-54, 2001.
- Yock TI, Krailo M, Fryer CJ, et al.: Local control in pelvic Ewing sarcoma: analysis from INT-0091--a report from the Children's Oncology Group. J Clin Oncol 24 (24): 3838-43, 2006.
- Schlegel M, Zeumer M, Prodinger PM, et al.: Impact of Pathological Fractures on the Prognosis of Primary Malignant Bone Sarcoma in Children and Adults: A Single-Center Retrospective Study of 205 Patients. Oncology 94 (6): 354-362, 2018.
- Bramer JA, Abudu AA, Grimer RJ, et al.: Do pathological fractures influence survival and local recurrence rate in bony sarcomas? Eur J Cancer 43 (13): 1944-51, 2007.
- Krasin MJ, Rodriguez-Galindo C, Billups CA, et al.: Definitive irradiation in multidisciplinary management of localized Ewing sarcoma family of tumors in pediatric patients: outcome and prognostic factors. Int J Radiat Oncol Biol Phys 60 (3): 830-8, 2004.
- Gerrand C, Bate J, Seddon B, et al.: Seeking international consensus on approaches to primary tumour treatment in Ewing sarcoma. Clin Sarcoma Res 10 (1): 21, 2020.
- Laskar S, Sinha S, Chatterjee A, et al.: Radiation Therapy Dose Escalation in Unresectable Ewing Sarcoma: Final Results of a Phase 3 Randomized Controlled Trial. Int J Radiat Oncol Biol Phys 113 (5): 996-1002, 2022.
- Kacar M, Nagel MB, Liang J, et al.: Radiation therapy dose escalation achieves high rates of local control with tolerable toxicity profile in pediatric and young adult patients with Ewing sarcoma. Cancer 130 (10): 1836-1843, 2024.
- Rombi B, DeLaney TF, MacDonald SM, et al.: Proton radiotherapy for pediatric Ewing's sarcoma: initial clinical outcomes. Int J Radiat Oncol Biol Phys 82 (3): 1142-8, 2012.
- Fuchs B, Valenzuela RG, Sim FH: Pathologic fracture as a complication in the treatment of Ewing's sarcoma. Clin Orthop (415): 25-30, 2003.
- Ahmed SK, Witten BG, Harmsen WS, et al.: Analysis of Local Control Outcomes and Clinical Prognostic Factors in Localized Pelvic Ewing Sarcoma Patients Treated With Radiation Therapy: A Report From the Children's Oncology Group. Int J Radiat Oncol Biol Phys 115 (2): 337-346, 2023.
- Schuck A, Ahrens S, Konarzewska A, et al.: Hemithorax irradiation for Ewing tumors of the chest wall. Int J Radiat Oncol Biol Phys 54 (3): 830-8, 2002.
- Kuttesch JF, Wexler LH, Marcus RB, et al.: Second malignancies after Ewing's sarcoma: radiation dose-dependency of secondary sarcomas. J Clin Oncol 14 (10): 2818-25, 1996.
- Whelan J, Le Deley MC, Dirksen U, et al.: High-Dose Chemotherapy and Blood Autologous Stem-Cell Rescue Compared With Standard Chemotherapy in Localized High-Risk Ewing Sarcoma: Results of Euro-E.W.I.N.G.99 and Ewing-2008. J Clin Oncol : JCO2018782516, 2018.
- Dirksen U, Brennan B, Le Deley MC, et al.: High-Dose Chemotherapy Compared With Standard Chemotherapy and Lung Radiation in Ewing Sarcoma With Pulmonary Metastases: Results of the European Ewing Tumour Working Initiative of National Groups, 99 Trial and EWING 2008. J Clin Oncol 37 (34): 3192-3202, 2019.
- Le Deley MC, Paulussen M, Lewis I, et al.: Cyclophosphamide compared with ifosfamide in consolidation treatment of standard-risk Ewing sarcoma: results of the randomized noninferiority Euro-EWING99-R1 trial. J Clin Oncol 32 (23): 2440-8, 2014.
Treatment of Metastatic Ewing Sarcoma
Approximately 25% of patients with Ewing sarcoma have metastases at diagnosis.[1] The prognosis is poor for patients with metastatic disease. With current therapies, patients who present with metastatic disease have a 6-year event-free survival (EFS) rate of approximately 28% and an overall survival (OS) rate of approximately 30%.[2,3] For patients with lung/pleural metastases only, the 6-year EFS rate is approximately 40% when using bilateral lung irradiation.[2,4] In contrast, patients with bone/bone marrow metastases have a 4-year EFS rate of approximately 28%, and patients with combined lung and bone/bone marrow metastases have a 4-year EFS rate of approximately 14%.[4,5]
The following factors independently predict a poor outcome in patients presenting with metastatic disease:[3]
- Age older than 14 years.
- Primary tumor volume of more than 200 mL.
- More than one bone metastatic site.
- Bone marrow metastases.
- Additional lung metastases.
- Pulmonary metastases lack of complete response to induction chemotherapy.[6]
Standard treatment options for metastatic Ewing sarcoma include the following:
- Chemotherapy.
- Surgery.
- Radiation therapy.
Chemotherapy
For patients with metastatic Ewing sarcoma, standard treatment that uses alternating cycles of vincristine/doxorubicin/cyclophosphamide and ifosfamide/etoposide (VDC/IE) combined with adequate local-control measures applied to both primary and metastatic sites often results in complete or partial responses. However, the overall cure rate is 20%.[5,7,8]
The following chemotherapy regimens have not shown benefit:
- In the Intergroup Ewing Sarcoma Study, patients with metastatic disease showed no benefit from the addition of ifosfamide and etoposide to a standard regimen of vincristine, doxorubicin, cyclophosphamide, and dactinomycin.[8]
- In another Intergroup study, increasing dose intensity of cyclophosphamide, ifosfamide, and doxorubicin did not improve outcome compared with regimens using standard-dose intensity. This regimen increased toxicity and risk of second malignancy without improving EFS or OS.[2]
- Intensification of ifosfamide to 2.8 g/m2 per day for 5 days did not improve outcome when administered with standard chemotherapy in patients with newly diagnosed metastatic Ewing sarcoma.[9][Level of evidence C2]
- A pilot study of low-dose anti-angiogenic therapy with vinblastine and celecoxib added to every-3-week therapy with VDC/IE did not improve outcomes for patients presenting with metastases.[10]
- The Children's Oncology Group performed a prospective randomized trial (AEWS1221 [NCT02306161]) that tested the addition of ganitumab to multiagent chemotherapy. Patients were randomly assigned (1:1) at enrollment to either the standard arm (interval-compressed VDC/IE) or the experimental arm (ganitumab is given with VDC/IE when the cycle starts and as monotherapy once every 3 weeks for 6 months after conventional therapy). The study enrolled 298 eligible patients (148 in standard arm; 150 in experimental arm).[11][Level of evidence B1]
- The 3-year EFS rate estimates were 37.4% (95% confidence interval [CI], 29.3%–45.5%) for patients in the standard arm and 39.1% (95% CI, 31.3%–46.7%) for patients in the experimental arm (stratified EFS-event hazard ratio [HR] for experimental arm, 1.00; 95% CI, 0.76–1.33; 1-sided, P = .50).
- Patients in the experimental arm reported more cases of pneumonitis after radiation therapy involving thoracic fields and nominally higher rates of febrile neutropenia and alanine transaminase elevation.
- Ganitumab added to interval-compressed chemotherapy did not significantly reduce the risk of an EFS event in patients with newly diagnosed metastatic Ewing sarcoma. The outcomes of these patients were similar to those in previous trials who did not receive IGF-1R inhibition or interval compression.
- The addition of ganitumab may be associated with increased toxicity.
Surgery and Radiation Therapy
Systematic use of surgery and radiation therapy for metastatic sites may improve overall outcome in patients with extrapulmonary metastases, although a randomized trial has not been done.
Evidence (surgery and radiation therapy):
- In a retrospective data analysis of 120 patients with multifocal metastatic Ewing sarcoma, patients who received local treatment to both the primary tumor and metastases had better outcomes than patients who received local treatment to the primary tumor only or with no local treatment (3-year EFS rate, 39% vs. 17% and 14%, respectively; P < .001).[12]
- A similar trend for better outcomes with irradiation of all sites of metastatic disease was seen in three retrospective analyses of smaller groups of patients who received radiation therapy to all tumor sites.[13,14,15]
These results must be interpreted with caution. The patients who received local-control therapy to all known sites of metastatic disease were selected by the treating investigator, not randomly assigned. Patients with so many metastases that radiation to all sites would result in bone marrow failure were not selected to receive radiation to all sites of metastatic disease. Patients who did not achieve control of the primary tumor did not go on to have local control of all sites of metastatic disease. There was a selection bias. While all patients in these reports had multiple sites of metastatic disease, the patients who had surgery and/or radiation therapy to all sites of clinically detectable metastatic disease had better responses to systemic therapy and fewer sites of metastasis than patients who did not undergo similar therapy of metastatic sites.
Radiation therapy, delivered in a setting in which stringent planning techniques are applied by those experienced in the treatment of Ewing sarcoma, should be considered. Such an approach will result in local control of the tumor with acceptable morbidity in most patients.[16]
The radiation dose depends on the metastatic site of disease:
- Bone and soft tissue. Stereotactic body radiation therapy has been used to treat metastatic sites in bone and soft tissue. The median total curative/definitive stereotactic body radiation therapy dose delivered was 40 Gy in five fractions (range, 30–60 Gy in 3–10 fractions). The median total palliative stereotactic body radiation therapy dose delivered was 40 Gy in five fractions (range, 16–50 Gy in 1–10 fractions). These short-course regimens with large-dose fractions are biologically equivalent to higher doses delivered with smaller-dose fractions given over longer treatment courses.[17][Level of evidence C1]
- Pulmonary. For all patients with pulmonary metastases, whole-lung irradiation should be considered, even if complete resolution of overt pulmonary metastatic disease has been achieved with chemotherapy.[4,5,18] Radiation doses are modulated based on age, the amount of lung to be irradiated, and pulmonary function. Doses between 12 Gy and 15 Gy are generally used if whole lungs are treated. No randomized trial has been done to prove radiation therapy improves survival in this group of patients. An early trial from the intergroup Ewing sarcoma group in the 1970s showed that radiation to the lung improved survival in patients with nonmetastatic disease when added to vincristine, actinomycin, and cyclophosphamide (VAC) compared with VAC alone.[19] However, the addition of doxorubicin to the therapy produced better EFS than the VAC/radiation therapy regimen.
Other Therapies
More intensive therapies, many of which incorporate high-dose chemotherapy with or without total-body irradiation in conjunction with stem cell support, have not improved EFS rates for patients with bone and/or bone marrow metastases.[2,3,13,20,21,22]; [23][Level of evidence C2] For more information, see the High-Dose Therapy With Stem Cell Support for Ewing Sarcoma section.
- High-dose chemotherapy with stem cell support.
- Ewing 2008R3 (NCT00987636) was the first phase III, open-label, multicenter, randomized controlled trial conducted for newly diagnosed patients with disseminated Ewing sarcoma who had metastases to bone and/or other sites.[24][Level of evidence B1] The study evaluated the EFS and OS effect of treatment with treosulfan and melphalan high-dose chemotherapy (TreoMel-HDT) followed by infusion of autologous hematopoietic stem cells. After six cycles of chemotherapy (vincristine, ifosfamide, doxorubicin, and etoposide), consenting patients were randomly assigned to receive either eight cycles of consolidation therapy (vincristine, dactinomycin, and cyclophosphamide) or consolidation therapy with TreoMel-HDT followed by autologous stem cell reinfusion. Between 2009 and 2018, 109 patients were randomly assigned, 55 of whom received TreoMel-HDT. There was no significant difference in 3-year EFS rates between patients who received TreoMel-HDT and patients in the control arm (20.9% vs. 19.2%; median follow-up, 3.3 years).
- One of the largest studies was the EURO-EWING-INTERGROUP-EE99 R3 trial that enrolled 281 patients with primary disseminated metastatic Ewing sarcoma. Patients were treated with six cycles of vincristine, ifosfamide, doxorubicin, and etoposide followed by high-dose therapy and autologous stem cell transplant. Patients had a 3-year EFS rate of 27% and an OS rate of 34%. Identified independent prognostic factors included the presence and number of bone lesions, primary tumor volume greater than 200 mL, age older than 14 years, additional pulmonary metastases, and bone marrow involvement.[3][Level of evidence C2]
- The role of high-dose therapy with busulfan-melphalan (BuMel) followed by stem cell rescue was investigated in the prospective randomized EURO-EWING-INTERGROUP-EE99 (NCT00020566) trial. Among patients with isolated pulmonary metastases, there was no difference in 3-year EFS rates (55.7% with BuMel vs. 50.3% with continued chemotherapy and whole-lung radiation therapy; P = .21).[25]
- Melphalan. At nonmyeloablative doses, melphalan proved to be an active agent in an up-front window study for patients with metastatic disease at diagnosis. However, the cure rate remained extremely low.[26]
- Irinotecan. Irinotecan was administered as a single agent in an up-front window study for patients with newly diagnosed metastatic Ewing sarcoma and showed modest activity (partial response in 5 of 24 patients).[27][Level of evidence C3] Further investigation is needed to determine irinotecan dosing and combinations with other agents for patients with Ewing sarcoma.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
- Esiashvili N, Goodman M, Marcus RB: Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. J Pediatr Hematol Oncol 30 (6): 425-30, 2008.
- Miser JS, Goldsby RE, Chen Z, et al.: Treatment of metastatic Ewing sarcoma/primitive neuroectodermal tumor of bone: evaluation of increasing the dose intensity of chemotherapy--a report from the Children's Oncology Group. Pediatr Blood Cancer 49 (7): 894-900, 2007.
- Ladenstein R, Pötschger U, Le Deley MC, et al.: Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol 28 (20): 3284-91, 2010.
- Paulussen M, Ahrens S, Craft AW, et al.: Ewing's tumors with primary lung metastases: survival analysis of 114 (European Intergroup) Cooperative Ewing's Sarcoma Studies patients. J Clin Oncol 16 (9): 3044-52, 1998.
- Paulussen M, Ahrens S, Burdach S, et al.: Primary metastatic (stage IV) Ewing tumor: survival analysis of 171 patients from the EICESS studies. European Intergroup Cooperative Ewing Sarcoma Studies. Ann Oncol 9 (3): 275-81, 1998.
- Halalsheh H, Kaste SC, Krasin MJ, et al.: Clinical impact of post-induction resolution of pulmonary lesions in metastatic Ewing sarcoma. Pediatr Blood Cancer 67 (4): e28150, 2020.
- Pinkerton CR, Bataillard A, Guillo S, et al.: Treatment strategies for metastatic Ewing's sarcoma. Eur J Cancer 37 (11): 1338-44, 2001.
- Miser JS, Krailo MD, Tarbell NJ, et al.: Treatment of metastatic Ewing's sarcoma or primitive neuroectodermal tumor of bone: evaluation of combination ifosfamide and etoposide--a Children's Cancer Group and Pediatric Oncology Group study. J Clin Oncol 22 (14): 2873-6, 2004.
- Magnan H, Goodbody CM, Riedel E, et al.: Ifosfamide dose-intensification for patients with metastatic Ewing sarcoma. Pediatr Blood Cancer 62 (4): 594-7, 2015.
- Felgenhauer JL, Nieder ML, Krailo MD, et al.: A pilot study of low-dose anti-angiogenic chemotherapy in combination with standard multiagent chemotherapy for patients with newly diagnosed metastatic Ewing sarcoma family of tumors: A Children's Oncology Group (COG) Phase II study NCT00061893. Pediatr Blood Cancer 60 (3): 409-14, 2013.
- DuBois SG, Krailo MD, Glade-Bender J, et al.: Randomized Phase III Trial of Ganitumab With Interval-Compressed Chemotherapy for Patients With Newly Diagnosed Metastatic Ewing Sarcoma: A Report From the Children's Oncology Group. J Clin Oncol 41 (11): 2098-2107, 2023.
- Haeusler J, Ranft A, Boelling T, et al.: The value of local treatment in patients with primary, disseminated, multifocal Ewing sarcoma (PDMES). Cancer 116 (2): 443-50, 2010.
- Burdach S, Thiel U, Schöniger M, et al.: Total body MRI-governed involved compartment irradiation combined with high-dose chemotherapy and stem cell rescue improves long-term survival in Ewing tumor patients with multiple primary bone metastases. Bone Marrow Transplant 45 (3): 483-9, 2010.
- Paulino AC, Mai WY, Teh BS: Radiotherapy in metastatic ewing sarcoma. Am J Clin Oncol 36 (3): 283-6, 2013.
- Casey DL, Wexler LH, Meyers PA, et al.: Radiation for bone metastases in Ewing sarcoma and rhabdomyosarcoma. Pediatr Blood Cancer 62 (3): 445-9, 2015.
- Donaldson SS, Torrey M, Link MP, et al.: A multidisciplinary study investigating radiotherapy in Ewing's sarcoma: end results of POG #8346. Pediatric Oncology Group. Int J Radiat Oncol Biol Phys 42 (1): 125-35, 1998.
- Brown LC, Lester RA, Grams MP, et al.: Stereotactic body radiotherapy for metastatic and recurrent ewing sarcoma and osteosarcoma. Sarcoma 2014: 418270, 2014.
- Spunt SL, McCarville MB, Kun LE, et al.: Selective use of whole-lung irradiation for patients with Ewing sarcoma family tumors and pulmonary metastases at the time of diagnosis. J Pediatr Hematol Oncol 23 (2): 93-8, 2001.
- Nesbit ME, Perez CA, Tefft M, et al.: Multimodal therapy for the management of primary, nonmetastatic Ewing's sarcoma of bone: an Intergroup Study. Natl Cancer Inst Monogr (56): 255-62, 1981.
- Meyers PA, Krailo MD, Ladanyi M, et al.: High-dose melphalan, etoposide, total-body irradiation, and autologous stem-cell reconstitution as consolidation therapy for high-risk Ewing's sarcoma does not improve prognosis. J Clin Oncol 19 (11): 2812-20, 2001.
- Burdach S, Meyer-Bahlburg A, Laws HJ, et al.: High-dose therapy for patients with primary multifocal and early relapsed Ewing's tumors: results of two consecutive regimens assessing the role of total-body irradiation. J Clin Oncol 21 (16): 3072-8, 2003.
- Thiel U, Wawer A, Wolf P, et al.: No improvement of survival with reduced- versus high-intensity conditioning for allogeneic stem cell transplants in Ewing tumor patients. Ann Oncol 22 (7): 1614-21, 2011.
- Loschi S, Dufour C, Oberlin O, et al.: Tandem high-dose chemotherapy strategy as first-line treatment of primary disseminated multifocal Ewing sarcomas in children, adolescents and young adults. Bone Marrow Transplant 50 (8): 1083-8, 2015.
- Koch R, Gelderblom H, Haveman L, et al.: High-Dose Treosulfan and Melphalan as Consolidation Therapy Versus Standard Therapy for High-Risk (Metastatic) Ewing Sarcoma. J Clin Oncol 40 (21): 2307-2320, 2022.
- Whelan J, Le Deley MC, Dirksen U, et al.: High-Dose Chemotherapy and Blood Autologous Stem-Cell Rescue Compared With Standard Chemotherapy in Localized High-Risk Ewing Sarcoma: Results of Euro-E.W.I.N.G.99 and Ewing-2008. J Clin Oncol : JCO2018782516, 2018.
- Luksch R, Grignani G, Fagioli F, et al.: Response to melphalan in up-front investigational window therapy for patients with metastatic Ewing's family tumours. Eur J Cancer 43 (5): 885-90, 2007.
- Morland B, Platt K, Whelan JS: A phase II window study of irinotecan (CPT-11) in high risk Ewing sarcoma: a Euro-E.W.I.N.G. study. Pediatr Blood Cancer 61 (3): 442-5, 2014.
Treatment of Recurrent Ewing Sarcoma
Recurrence of Ewing sarcoma is most common within 2 years of initial diagnosis (approximately 80%).[1,2] However, late relapses occurring more than 5 years from initial diagnosis are more common in Ewing sarcoma (13%; 95% confidence interval, 9.4%–16.5%) than in other pediatric solid tumors.[3] An analysis of the Surveillance, Epidemiology, and End Results (SEER) Program database identified 1,351 patients who survived more than 60 months from diagnosis.[4] Of these patients, 209 died; 144 of the deaths (69%) were attributed to recurrent, progressive Ewing sarcoma. Black race, male sex, older age at initial diagnosis, and primary tumors of the pelvis and axial skeleton were associated with a higher risk of late death. This analysis covered the period from 1973 to 2013, and the 1,351 patients represented only 38% of the patients in the original sample, which reflects the inferior treatment outcomes from the earlier era. It is possible that patients who reach the 5-year point after more contemporary treatment may not recapitulate this experience.
The overall prognosis for patients with recurrent Ewing sarcoma is poor. The 5-year survival rate after recurrence is approximately 10% to 15%.[2,5,6]; [1][Level of evidence C1] Patients with relapsed or progressive Ewing sarcoma with measurable disease have a 6-month event-free survival (EFS) rate of 13%.[7][Level of evidence C1]
Prognostic factors include the following:
- Time to recurrence. Time to recurrence is the most important prognostic factor. Patients whose Ewing sarcoma recurred more than 2 years from initial diagnosis had a 5-year survival rate of 30%, versus 7% for patients whose disease recurred within 2 years.[1,2]
- Local and distant recurrence. Patients with both local recurrence and distant metastases have a worse outcome than patients with either isolated local recurrence or metastatic recurrence alone.[1,2]
- Isolated pulmonary recurrence. Isolated pulmonary recurrence was not an important prognostic factor in a North American series.[1] In the Italian/Scandinavian experience, younger age, longer disease-free interval, and lung-only recurrence were associated with longer progression-free survival (PFS) after recurrence. In this experience, patients with Ewing sarcoma that recurred after initial therapy, which included high-dose therapy with autologous stem cell rescue, were less likely to achieve a second complete remission.[8][Level of evidence C2]
The selection of treatment for patients with recurrent disease depends on many factors, including the following:
- Site of recurrence.
- Previous treatment.
- Individual patient considerations.
There is no standardized second-line treatment for patients with relapsed or refractory Ewing sarcoma. Most patients in first relapse are treated with conventional systemic chemotherapy. Patients who demonstrate a response to therapy may undergo local control to sites of recurrence.
Treatment options for recurrent Ewing sarcoma include the following:
- Chemotherapy.
- Surgery.
- Radiation therapy.
- High-dose chemotherapy with stem cell support.
- Other therapies.
Chemotherapy
Combinations of chemotherapy, such as cyclophosphamide and topotecan or irinotecan and temozolomide with or without vincristine, are active in recurrent Ewing sarcoma and can be considered for these patients.[9,10,11,12,13,14]
Table 7. Results from Studies that Used Cyclophosphamide and Topotecan Regimens to Treat Patients With Relapsed and/or Refractory Ewing Sarcoma| Study Reference | Trial Phase (Total No. of Patients) | Median Age (Range) (y) | CR/PR | RR | Cyclophosphamide (mg/m2)/Topotecan (mg/m2) × d | Other Agents |
|---|
| II = phase II trial; CR = complete response; PR = partial response; R = retrospective; RR = objective response rate; VCR = vincristine. |
| Saylors et al.[9] | II (17) | 13.8 (1–21) | 1/3 | 29% | 250 × 5/0.75 × 5 | None |
| Hunold et al.[11] | R (54) | 17.4 (3–49) | 0/16 | 30% | 250 × 5/0.75 × 5 | None |
| Farhat et al.[15] | R (14) | 11 (2–19) | 0/3 | 21% | 250 × 5/0.75 × 5 | None |
| Kebudi et al.[16] | R (14) | 13 (3–16) | 2/5 | 50% | 250 × 5/0.75 × 5 | VCR |
These studies were retrospective. Prospective trials with clearly defined eligibility cohorts and intent-to-treat analyses are lacking. When combined, these studies accrued 99 patients and observed 3 complete remissions and 27 partial remissions. The objective response rate was 30%.
Table 8. Results from Studies that Used Temozolomide and Irinotecan Regimens to Treat Patients With Relapsed and/or Refractory Ewing Sarcoma| Study Reference | Trial Phase (Total No. of Patients) | Median Age (Range) (y) | CR/PR | RR | Temozolomide (mg/m2)/Irinotecan (mg/m2) × d × wk | Other Agents |
|---|
| I = phase I trial; II = phase II trial; Acta Onc = Acta Oncologica; An Ped = Annals of Pediatrics; Clin Cancer Res = Clinical Cancer Research; Exp Opin = Expert Opinion Investigational Drugs; Clin Transl Oncol = Clinical and Translational Oncology; BEV = bevacizumab; CR = complete response; IV = intravenous; N/A = not applicable; PBC = Pediatric Blood and Cancer; Ped Hem Onc = Pediatric Hematology and Oncology; PO = oral; PR = partial response; R = retrospective trial; RR = objective response rate; TMS = temsirolimus; UK = unknown; VCR = vincristine. |
| Wagner et al. (PBC, 2007)[12] | R (16) | 18 (7–33) | 1/3 | 29% | 100 × 5/IV 10–20 × 5 × 2 | None |
| Casey et al. (PBC, 2009)[13] | R (19) | 19.5 (2–40) | 5/7 | 63% | 100 × 5/IV 20 × 5 × 2 | None |
| Hernandez-Marques et al. (An Ped, 2013)[17] | R (8) | 13 (6–18) | 0/3 | 37% | 80–100 × 5/IV 10–20 × 5 × 2 | None |
| Raciborska et al. (PBC, 2013)[14] | R (22) | 14.3 | 5/7 | 54% | 125 × 5/IV 50 × 5 | VCR |
| McKnall-Knapp et al. (PBC, 2010)[18] | I (1) | N/A | 0/1 | 100% | 100 × 5/IV 20 × 5 × 2 | VCR |
| Wagner et al. (PBC, 2010)[19] | I (5) | (<21) | 1/1 | 40% | 100–150 × 5/PO 35–90 × 5 | VCR |
| Wagner et al. (PBC, 2013)[20] | I (2) | 20, 22 | 1/1 | 100% | 150 × 5/PO 90 × 5 | VCR, BEV |
| Bagatell et al. (PBC, 2014)[21] | I (7) | (<21) | 0/1 | 14% | 100–150 × 5/PO 50–90 × 5 | TMS |
| Kurucu et al. (Ped Hem Onc, 2015)[22] | R (20) | 14 (1–18) | UK | 55% | 100 × 5/IV 20 × 5 × 2 | None |
| Anderson et al. (Exp Opin, 2008)[23] | R (25) | 15 | 7/9 | 64% | 100 × 5/IV 10 × 5 × 2 | None |
| Palmerini et al. (Acta Onc, 2018)[24] | R (51) | 21 (3–65) | 5/12 | 34% | 100 × 5/IV 40 × 5 | None |
| Salah et al. (Clin Transl Oncol, 2021)[25] | R (53) | 20 (5–45) | 1/11 | 28% | 100 × 5/IV 40 × 5 in 21 patients; IV 50 × 5 in 24 patients; IV 20 × 5 × 2 in 6 patients | None |
| Xu et al. (Clin Cancer Res, 2023)[26]5-day schedule: | II (24) | 16.5 ± 7.9 | 1/4 | 20.8% | 100 × 5/50 × 5 | VCR 1.4 mg/m2 day 1 |
| Xu et al. (Clin Cancer Res, 2023)[26]10-day schedule: | II (22) | 15.2 ± 6.3 | 1/11 | 54.5% | 100 × 5/20 × 5 × 2 | VCR 1.4 mg/m2 days 1 and 8 |
Most of these studies were retrospective, not prospective. There are only four prospective trials with well-defined eligibility cohorts and report by intent to treat. In addition, there is significant variability among the reports in doses and dose schedules of irinotecan and temozolomide and the use of additional agents. When combined, these studies accrued 275 patients and observed 21 complete remissions and 82 partial remissions. The objective response rate was 37.5%.
Evidence (chemotherapy):
- One phase II study of topotecan and cyclophosphamide showed a response in 6 of 17 patients with Ewing sarcoma.[9] In a similar trial in Germany, 16 of 49 patients had a clinical response.[11]
- Several retrospective studies have demonstrated the activity of temozolomide and irinotecan in patients with recurrent Ewing sarcoma.[13,22,24]
- In the largest retrospective multicenter study of the combination of temozolomide and irinotecan in patients with recurrent and primary refractory Ewing sarcoma, 51 patients (66% of patients were aged ≥18 years; median age, 21 years) were treated with temozolomide (100 mg/m2 /day orally) and irinotecan (40 mg/m2 /day intravenously), on days 1 to 5, every 21 days. Twenty-five percent of the patients were in first relapse/progression, while the remainder of the patients were in second or greater relapse/progression.[24]
- Five patients (10%) achieved complete remissions, 12 patients (24%) achieved partial remissions, and 19 patients (37%) had stable disease, with a disease control rate of 71%.
- On univariate analysis, the only two factors predicting response to temozolomide and irinotecan in PFS were performance score and lactate dehydrogenase levels.
- Two patients were rechallenged with temozolomide and irinotecan after disease remission was induced. Both patients achieved partial remissions on rechallenge. One patient's remission lasted at least 15 cycles and the other remission lasted 22 cycles.[24]
- A prospective randomized trial compared two schedules of irinotecan given in combination with vincristine and temozolomide for the treatment of recurrent Ewing sarcoma. One schedule administered irinotecan 50 mg/m2 daily for 5 days (d × 5) and the other administered irinotecan 20 mg/m2 daily for 5 days for two consecutive weeks (10 doses; d × 5 × 2).[26]
- The objective response rate at 12 weeks was lower for the patients treated on the d × 5 schedule (5 of 24; 20.8%) than for patients treated on the d × 5 × 2 schedule (12 of 22; 54.5%; P = .019).
- A retrospective review of published series compared the results of treatment with irinotecan and temozolomide with a 10-day schedule to treatment with a 5-day schedule.[27]
- Among 89 patients treated with a 10-day irinotecan schedule, there were 47 objective responses (53%).
- Among 180 patients treated with a 5-day irinotecan schedule, there were 52 responses (29%).
- The two studies that used the 5-day irinotecan schedule reported median times to progression of 3.0 and 3.9 months, respectively.
- The four studies that used the 10-day irinotecan schedule reported median times to progression of 4.6, 5.5, 8.3, and 9.5 months, respectively.
- The combination of docetaxel either with gemcitabine or irinotecan has achieved objective responses in patients with relapsed Ewing sarcoma.[28][Level of evidence C1]; [29,30][Level of evidence C3]
- High-dose ifosfamide (3 g/m2 per day for 5 days = 15 g/m2) has shown activity in patients whose Ewing sarcoma recurred after therapy that included standard ifosfamide (1.8 g/m2 per day for 5 days = 9 g/m2).[31][Level of evidence C3]
- European investigators are performing a prospective study to compare four regimens for the treatment of patients with recurrent Ewing sarcoma.[32] The rEECur phase II/III adaptive multiarm trial is the first to compare regimens in a randomized design. Patients aged 4 to 50 years with refractory or recurrent Ewing sarcoma and healthy enough to receive chemotherapy were randomly assigned to receive one of four regimens: topotecan and cyclophosphamide (TC), irinotecan and temozolomide (IT), gemcitabine and docetaxel (GD), or high-dose ifosfamide.[33]
- Pairwise comparison showed that GD was the least effective.
- Imaging response and survival outcomes after TC were marginally better than after IT. However, in a phase II comparison, ifosfamide had significantly better PFS and OS than TC. As a result, ifosfamide was the most effective regimen in this setting, although with significant renal and neurological toxicity.
- The study is ongoing and has recruited over 570 patients. The study is now comparing ifosfamide and lenvatinib with ifosfamide, carboplatin, and etoposide.[34]
Local Therapy for Relapsed Disease
Surgery
Aggressive surgery (such as amputation or hemipelvectomy) may be considered for patients with nonmetastatic locally recurrent Ewing sarcoma, even if the prognosis is limited.[35]
The role of pulmonary metastasectomy in patients with relapsed disease and isolated lung metastases is controversial.[36,37]
Radiation therapy
Radiation therapy may be used (similar to first-line strategies) for patients who relapsed after the beginning of front-line therapy and/or who present only with relapsed pulmonary metastases.[36]; [38][Level of evidence C1] Radiation therapy to bone lesions may provide palliation, although radical resection may improve outcome.[2] Patients with pulmonary metastases who have not received radiation therapy to the lungs should be considered for whole-lung irradiation and/or treated with stereotactic body radiation therapy.[36,39]; [38][Level of evidence C1]; Residual disease in the lung may be surgically removed.
Palliation of painful lesions in children with recurrent or progressive disease can be achieved using a short course (10 or fewer fractions) of radiation therapy. In a retrospective study of 213 children with various malignancies, who were treated with such short course radiation therapy, 85% of patients had complete or partial pain relief, with low levels of toxicity.[40]
High-Dose Chemotherapy With Stem Cell Support
Aggressive attempts to control the disease, including myeloablative regimens, have been used, but there is no evidence at this time to conclude that myeloablative therapy is superior to standard chemotherapy.[41,42,43]; [44][Level of evidence C2]
Most published reports about the use of high-dose therapy and stem cell support for patients with high-risk Ewing sarcoma have significant flaws in methodology. The most common issue is the comparison of this high-risk group with an inappropriate control group. Patients with Ewing sarcoma at high risk of treatment failure who received high-dose therapy are compared with patients who did not receive high-dose therapy. Patients who undergo high-dose therapy must respond to systemic therapy, remain alive and respond to treatment long enough to reach the time at which stem cell therapy can be applied, be free of comorbid toxicity that precludes high-dose therapy, and have an adequate stem cell collection. Patients who undergo high-dose therapy and stem cell support are a highly selected group. Comparing this patient group with all patients with high-risk Ewing sarcoma is inappropriate and leads to the erroneous conclusion that this strategy improves outcome.
Surveys of patients who underwent allogeneic hematopoietic stem cell transplant (HSCT) for recurrent Ewing sarcoma did not show improved EFS when compared with patients who underwent autologous HSCT. In addition, allogeneic HSCT was associated with a higher complication rate.[41,45,46]
Other Therapies
Other therapies that have been studied in the treatment of recurrent Ewing sarcoma include the following:
Sequencing of recurrent and refractory Ewing sarcoma tumors from pediatric (n = 79) and young adult patients (n = 25) enrolled in the National Cancer Institute (NCI)-COG Pediatric Molecular Analysis for Therapeutic Choice (MATCH) trial revealed genomic alterations that were considered actionable for treatment on MATCH study arms in 8 of 104 tumors (7.7%), including EZH2 variants in 2 of 104 tumors (1.9%).[71]
Treatment Options Under Clinical Evaluation for Recurrent Ewing Sarcoma
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- NCT04890093 (Vincristine and Temozolomide in Combination With PEN-866 for Adolescents and Young Adults With Relapsed or Refractory Solid Tumors): PEN-866 is a novel molecule consisting of SN-38 conjugated to a heat shock protein 90 (HSP90) inhibitor that has been shown to have a pharmacokinetic advantage over irinotecan in preclinical models. In preclinical models of Ewing sarcoma, PEN-866 had superior efficacy and pharmacodynamics compared with irinotecan. For the phase I portion of this trial, any patient aged 12 to 39 years with a relapsed or refractory solid tumor is eligible. The phase II portion of this trial will enroll only patients aged 12 to 39 years with relapsed or refractory Ewing sarcoma or rhabdomyosarcoma.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
- Leavey PJ, Mascarenhas L, Marina N, et al.: Prognostic factors for patients with Ewing sarcoma (EWS) at first recurrence following multi-modality therapy: A report from the Children's Oncology Group. Pediatr Blood Cancer 51 (3): 334-8, 2008.
- Stahl M, Ranft A, Paulussen M, et al.: Risk of recurrence and survival after relapse in patients with Ewing sarcoma. Pediatr Blood Cancer 57 (4): 549-53, 2011.
- Wasilewski-Masker K, Liu Q, Yasui Y, et al.: Late recurrence in pediatric cancer: a report from the Childhood Cancer Survivor Study. J Natl Cancer Inst 101 (24): 1709-20, 2009.
- Davenport JR, Vo KT, Goldsby R, et al.: Conditional Survival and Predictors of Late Death in Patients With Ewing Sarcoma. Pediatr Blood Cancer 63 (6): 1091-5, 2016.
- Barker LM, Pendergrass TW, Sanders JE, et al.: Survival after recurrence of Ewing's sarcoma family of tumors. J Clin Oncol 23 (19): 4354-62, 2005.
- Bacci G, Longhi A, Ferrari S, et al.: Pattern of relapse in 290 patients with nonmetastatic Ewing's sarcoma family tumors treated at a single institution with adjuvant and neoadjuvant chemotherapy between 1972 and 1999. Eur J Surg Oncol 32 (9): 974-9, 2006.
- Collier AB, Krailo MD, Dang HM, et al.: Outcome of patients with relapsed or progressive Ewing sarcoma enrolled on cooperative group phase 2 clinical trials: A report from the Children's Oncology Group. Pediatr Blood Cancer 68 (12): e29333, 2021.
- Ferrari S, Luksch R, Hall KS, et al.: Post-relapse survival in patients with Ewing sarcoma. Pediatr Blood Cancer 62 (6): 994-9, 2015.
- Saylors RL, Stine KC, Sullivan J, et al.: Cyclophosphamide plus topotecan in children with recurrent or refractory solid tumors: a Pediatric Oncology Group phase II study. J Clin Oncol 19 (15): 3463-9, 2001.
- McTiernan A, Driver D, Michelagnoli MP, et al.: High dose chemotherapy with bone marrow or peripheral stem cell rescue is an effective treatment option for patients with relapsed or progressive Ewing's sarcoma family of tumours. Ann Oncol 17 (8): 1301-5, 2006.
- Hunold A, Weddeling N, Paulussen M, et al.: Topotecan and cyclophosphamide in patients with refractory or relapsed Ewing tumors. Pediatr Blood Cancer 47 (6): 795-800, 2006.
- Wagner LM, McAllister N, Goldsby RE, et al.: Temozolomide and intravenous irinotecan for treatment of advanced Ewing sarcoma. Pediatr Blood Cancer 48 (2): 132-9, 2007.
- Casey DA, Wexler LH, Merchant MS, et al.: Irinotecan and temozolomide for Ewing sarcoma: the Memorial Sloan-Kettering experience. Pediatr Blood Cancer 53 (6): 1029-34, 2009.
- Raciborska A, Bilska K, Drabko K, et al.: Vincristine, irinotecan, and temozolomide in patients with relapsed and refractory Ewing sarcoma. Pediatr Blood Cancer 60 (10): 1621-5, 2013.
- Farhat R, Raad R, Khoury NJ, et al.: Cyclophosphamide and topotecan as first-line salvage therapy in patients with relapsed ewing sarcoma at a single institution. J Pediatr Hematol Oncol 35 (5): 356-60, 2013.
- Kebudi R, Cakir FB, Gorgun O, et al.: A modified protocol with vincristine, topotecan, and cyclophosphamide for recurrent/progressive ewing sarcoma family tumors. Pediatr Hematol Oncol 30 (3): 170-7, 2013.
- Hernández-Marqués C, Lassaletta-Atienza A, Ruiz Hernández A, et al.: [Irinotecan plus temozolomide in refractory or relapsed pediatric solid tumors]. An Pediatr (Barc) 79 (2): 68-74, 2013.
- McNall-Knapp RY, Williams CN, Reeves EN, et al.: Extended phase I evaluation of vincristine, irinotecan, temozolomide, and antibiotic in children with refractory solid tumors. Pediatr Blood Cancer 54 (7): 909-15, 2010.
- Wagner LM, Perentesis JP, Reid JM, et al.: Phase I trial of two schedules of vincristine, oral irinotecan, and temozolomide (VOIT) for children with relapsed or refractory solid tumors: a Children's Oncology Group phase I consortium study. Pediatr Blood Cancer 54 (4): 538-45, 2010.
- Wagner L, Turpin B, Nagarajan R, et al.: Pilot study of vincristine, oral irinotecan, and temozolomide (VOIT regimen) combined with bevacizumab in pediatric patients with recurrent solid tumors or brain tumors. Pediatr Blood Cancer 60 (9): 1447-51, 2013.
- Bagatell R, Norris R, Ingle AM, et al.: Phase 1 trial of temsirolimus in combination with irinotecan and temozolomide in children, adolescents and young adults with relapsed or refractory solid tumors: a Children's Oncology Group Study. Pediatr Blood Cancer 61 (5): 833-9, 2014.
- Kurucu N, Sari N, Ilhan IE: Irinotecan and temozolamide treatment for relapsed Ewing sarcoma: a single-center experience and review of the literature. Pediatr Hematol Oncol 32 (1): 50-9, 2015.
- Anderson P, Kopp L, Anderson N, et al.: Novel bone cancer drugs: investigational agents and control paradigms for primary bone sarcomas (Ewing's sarcoma and osteosarcoma). Expert Opin Investig Drugs 17 (11): 1703-15, 2008.
- Palmerini E, Jones RL, Setola E, et al.: Irinotecan and temozolomide in recurrent Ewing sarcoma: an analysis in 51 adult and pediatric patients. Acta Oncol 57 (7): 958-964, 2018.
- Salah S, To YH, Khozouz O, et al.: Irinotecan and temozolomide chemotherapy in paediatric and adult populations with relapsed Ewing Sarcoma. Clin Transl Oncol 23 (4): 757-763, 2021.
- Xu J, Xie L, Sun X, et al.: Longer versus Shorter Schedules of Vincristine, Irinotecan, and Temozolomide (VIT) for Relapsed or Refractory Ewing Sarcoma: A Randomized Controlled Phase 2 Trial. Clin Cancer Res 29 (6): 1040-1046, 2023.
- Slotkin EK, Meyers PA: Irinotecan dose schedule for the treatment of Ewing sarcoma. Pediatr Blood Cancer 70 (1): e30005, 2023.
- Fox E, Patel S, Wathen JK, et al.: Phase II study of sequential gemcitabine followed by docetaxel for recurrent Ewing sarcoma, osteosarcoma, or unresectable or locally recurrent chondrosarcoma: results of Sarcoma Alliance for Research Through Collaboration Study 003. Oncologist 17 (3): 321, 2012.
- Mora J, Cruz CO, Parareda A, et al.: Treatment of relapsed/refractory pediatric sarcomas with gemcitabine and docetaxel. J Pediatr Hematol Oncol 31 (10): 723-9, 2009.
- Yoon JH, Kwon MM, Park HJ, et al.: A study of docetaxel and irinotecan in children and young adults with recurrent or refractory Ewing sarcoma family of tumors. BMC Cancer 14: 622, 2014.
- Ferrari S, del Prever AB, Palmerini E, et al.: Response to high-dose ifosfamide in patients with advanced/recurrent Ewing sarcoma. Pediatr Blood Cancer 52 (5): 581-4, 2009.
- McCabe MG, Moroz V, Khan M, et al.: Results of the first interim assessment of rEECur, an international randomized controlled trial of chemotherapy for the treatment of recurrent and primary refractory Ewing sarcoma. [Abstract] J Clin Oncol 37 (Suppl 15): A-11007, 2019. Also available online. Last accessed June 29, 2021.
- McCabe MG, Kirton L, Khan M, et al.: Results of the second interim assessment of rEECur, an international randomized controlled trial of chemotherapy for the treatment of recurrent and primary refractory Ewing sarcoma (RR-ES). [Abstract] J Clin Oncol 38 (Suppl 15): A-11502, 2020. Also available online. Last accessed June 29, 2021.
- Strauss SJ, Berlanga P, McCabe MG: Emerging therapies in Ewing sarcoma. Curr Opin Oncol 36 (4): 297-304, 2024.
- Gerrand C, Bate J, Seddon B, et al.: Seeking international consensus on approaches to primary tumour treatment in Ewing sarcoma. Clin Sarcoma Res 10 (1): 21, 2020.
- Rodriguez-Galindo C, Billups CA, Kun LE, et al.: Survival after recurrence of Ewing tumors: the St Jude Children's Research Hospital experience, 1979-1999. Cancer 94 (2): 561-9, 2002.
- Briccoli A, Rocca M, Ferrari S, et al.: Surgery for lung metastases in Ewing's sarcoma of bone. Eur J Surg Oncol 30 (1): 63-7, 2004.
- Scobioala S, Ranft A, Wolters H, et al.: Impact of Whole Lung Irradiation on Survival Outcome in Patients With Lung Relapsed Ewing Sarcoma. Int J Radiat Oncol Biol Phys 102 (3): 584-592, 2018.
- Liu KX, Chen YH, Kozono D, et al.: Phase I/II Study of Stereotactic Body Radiation Therapy for Pulmonary Metastases in Pediatric Patients. Adv Radiat Oncol 5 (6): 1267-1273, 2020 Nov-Dec.
- Sudmeier LJ, Madden N, Zhang C, et al.: Palliative radiotherapy for children: Symptom response and treatment-associated toxicity according to radiation therapy dose and fractionation. Pediatr Blood Cancer 70 (4): e30195, 2023.
- Burdach S, van Kaick B, Laws HJ, et al.: Allogeneic and autologous stem-cell transplantation in advanced Ewing tumors. An update after long-term follow-up from two centers of the European Intergroup study EICESS. Stem-Cell Transplant Programs at Düsseldorf University Medical Center, Germany and St. Anna Kinderspital, Vienna, Austria. Ann Oncol 11 (11): 1451-62, 2000.
- Burdach S, Meyer-Bahlburg A, Laws HJ, et al.: High-dose therapy for patients with primary multifocal and early relapsed Ewing's tumors: results of two consecutive regimens assessing the role of total-body irradiation. J Clin Oncol 21 (16): 3072-8, 2003.
- Pawlowska AB, Sun V, Calvert GT, et al.: Long-Term Follow-up of High-Dose Chemotherapy with Autologous Stem Cell Transplantation in Children and Young Adults with Metastatic or Relapsed Ewing Sarcoma: A Single-Institution Experience. Transplant Cell Ther 27 (1): 72.e1-72.e7, 2021.
- Gardner SL, Carreras J, Boudreau C, et al.: Myeloablative therapy with autologous stem cell rescue for patients with Ewing sarcoma. Bone Marrow Transplant 41 (10): 867-72, 2008.
- Gilman AL, Oesterheld J: Myeloablative chemotherapy with autologous stem cell rescue for Ewing sarcoma. Bone Marrow Transplant 42 (11): 761; author reply 763, 2008.
- Eapen M: Response to Dr Gilman. Bone Marrow Transplant 42 (11): 763, 2008.
- Malempati S, Weigel B, Ingle AM, et al.: Phase I/II trial and pharmacokinetic study of cixutumumab in pediatric patients with refractory solid tumors and Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 30 (3): 256-62, 2012.
- Juergens H, Daw NC, Geoerger B, et al.: Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J Clin Oncol 29 (34): 4534-40, 2011.
- Pappo AS, Patel SR, Crowley J, et al.: R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a phase II Sarcoma Alliance for Research through Collaboration study. J Clin Oncol 29 (34): 4541-7, 2011.
- Tap WD, Demetri G, Barnette P, et al.: Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J Clin Oncol 30 (15): 1849-56, 2012.
- Shulman DS, Merriam P, Choy E, et al.: Phase 2 trial of palbociclib and ganitumab in patients with relapsed Ewing sarcoma. Cancer Med 12 (14): 15207-15216, 2023.
- Naing A, LoRusso P, Fu S, et al.: Insulin growth factor-receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with refractory Ewing's sarcoma family tumors. Clin Cancer Res 18 (9): 2625-31, 2012.
- Schwartz GK, Tap WD, Qin LX, et al.: Cixutumumab and temsirolimus for patients with bone and soft-tissue sarcoma: a multicentre, open-label, phase 2 trial. Lancet Oncol 14 (4): 371-82, 2013.
- Ahmed N, Brawley VS, Hegde M, et al.: Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol 33 (15): 1688-96, 2015.
- Pule MA, Savoldo B, Myers GD, et al.: Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 14 (11): 1264-70, 2008.
- Scotlandi K, Baldini N, Cerisano V, et al.: CD99 engagement: an effective therapeutic strategy for Ewing tumors. Cancer Res 60 (18): 5134-42, 2000.
- Grunewald TG, Diebold I, Esposito I, et al.: STEAP1 is associated with the invasive and oxidative stress phenotype of Ewing tumors. Mol Cancer Res 10 (1): 52-65, 2012.
- D'Angelo SP, Mahoney MR, Van Tine BA, et al.: Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol 19 (3): 416-426, 2018.
- Tawbi HA, Burgess M, Bolejack V, et al.: Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol 18 (11): 1493-1501, 2017.
- Davis KL, Fox E, Merchant MS, et al.: Nivolumab in children and young adults with relapsed or refractory solid tumours or lymphoma (ADVL1412): a multicentre, open-label, single-arm, phase 1-2 trial. Lancet Oncol 21 (4): 541-550, 2020.
- Davis KL, Fox E, Isikwei E, et al.: A Phase I/II Trial of Nivolumab plus Ipilimumab in Children and Young Adults with Relapsed/Refractory Solid Tumors: A Children's Oncology Group Study ADVL1412. Clin Cancer Res 28 (23): 5088-5097, 2022.
- Attia S, Bolejack V, Ganjoo KN, et al.: A phase II trial of regorafenib in patients with advanced Ewing sarcoma and related tumors of soft tissue and bone: SARC024 trial results. Cancer Med 12 (2): 1532-1539, 2023.
- Duffaud F, Blay JY, Le Cesne A, et al.: Regorafenib in patients with advanced Ewing sarcoma: results of a non-comparative, randomised, double-blind, placebo-controlled, multicentre Phase II study. Br J Cancer 129 (12): 1940-1948, 2023.
- Casanova M, Bautista F, Campbell-Hewson Q, et al.: Regorafenib plus Vincristine and Irinotecan in Pediatric Patients with Recurrent/Refractory Solid Tumors: An Innovative Therapy for Children with Cancer Study. Clin Cancer Res 29 (21): 4341-4351, 2023.
- Xu J, Xie L, Sun X, et al.: Anlotinib, Vincristine, and Irinotecan for Advanced Ewing Sarcoma After Failure of Standard Multimodal Therapy: A Two-Cohort, Phase Ib/II Trial. Oncologist 26 (7): e1256-e1262, 2021.
- Peretz Soroka H, Vora T, Noujaim J, et al.: Real-world experience of tyrosine kinase inhibitors in children, adolescents and adults with relapsed or refractory bone tumours: A Canadian Sarcoma Research and Clinical Collaboration (CanSaRCC) study. Cancer Med 12 (18): 18872-18881, 2023.
- Italiano A, Mir O, Mathoulin-Pelissier S, et al.: Cabozantinib in patients with advanced Ewing sarcoma or osteosarcoma (CABONE): a multicentre, single-arm, phase 2 trial. Lancet Oncol 21 (3): 446-455, 2020.
- Subbiah V, Braña I, Longhi A, et al.: Antitumor Activity of Lurbinectedin, a Selective Inhibitor of Oncogene Transcription, in Patients with Relapsed Ewing Sarcoma: Results of a Basket Phase II Study. Clin Cancer Res 28 (13): 2762-2770, 2022.
- Povedano JM, Li V, Lake KE, et al.: TK216 targets microtubules in Ewing sarcoma cells. Cell Chem Biol 29 (8): 1325-1332.e4, 2022.
- Meyers PA, Federman N, Daw N, et al.: Open-Label, Multicenter, Phase I/II, First-in-Human Trial of TK216: A First-Generation EWS::FLI1 Fusion Protein Antagonist in Ewing Sarcoma. J Clin Oncol 42 (31): 3725-3734, 2024.
- Parsons DW, Janeway KA, Patton DR, et al.: Actionable Tumor Alterations and Treatment Protocol Enrollment of Pediatric and Young Adult Patients With Refractory Cancers in the National Cancer Institute-Children's Oncology Group Pediatric MATCH Trial. J Clin Oncol 40 (20): 2224-2234, 2022.
Undifferentiated Small Round Cell (Ewing-Like) Sarcomas
There are undifferentiated small round cell sarcomas of bone and soft tissue that do not have the translocations of the EWSR1 gene and a gene in the ETS family. These sarcomas appear to be biologically distinctive from Ewing sarcoma with EWSR1 and ETS gene family member translocations. This includes tumors with translocations of the CIC gene or the BCOR gene, as well as tumors with EWSR1 translocations involving non-ETS gene family members. These groups occur much less frequently than Ewing sarcoma, and descriptions of clinical outcomes for these patients are based on smaller sample sizes and less homogeneous treatment. Therefore, patient outcomes are hard to quantitate with precision. Most of these tumors have been treated with regimens designed for Ewing sarcoma, and there is consensus that they were often included in past clinical trials for the treatment of Ewing sarcoma, sometimes called translocation-negative Ewing sarcoma. There is agreement that these tumors are sufficiently different from Ewing sarcoma; they should be stratified and analyzed separately from Ewing sarcoma with the common translocations, even if they are treated with similar therapy. The summary of these entities are presented below and follows the categorization of the 2020 World Health Organization (WHO) Classification of Tumours: Soft Tissue and Bone Tumours (5th edition).[1]
Undifferentiated Small Round Cell Sarcomas WithBCORGenetic Alterations
Clinical presentation
Undifferentiated round cell sarcomas with BCOR::CCNB3 rearrangements account for about 5% of all EWSR1-negative rearranged sarcomas and more commonly affects males. More than 70% of cases occur in patients younger than 18 years (median age at diagnosis, 13–15 years).[2,3][Level of evidence C1] These tumors more commonly arise in the bones of the pelvis and extremities, and metastases are present in approximately 30% of cases.
Genomic characteristics
The most common types of undifferentiated small round cell sarcoma with BCOR rearrangements are those with the BCOR::CCNB3 rearrangement.[2,4] The BCOR::MAML3 rearrangement is less commonly observed, but tumors with this translocation appear to have biological characteristics that are similar to tumors with the BCOR::CCNB3 rearrangement.[2,5,6]
BCOR internal tandem duplications (ITD) involving exon 15 are observed in infantile undifferentiated round cell sarcomas and primitive myxoid mesenchymal tumors of infancy (PMMTI).[7,8,9] These two entities have significant histological overlap and similar transcriptional profiles, and they are distinguished by more prominent myxoid stroma in PMMTI. BCOR ITD may be occasionally observed in undifferentiated round cell sarcomas arising in older children.[9]
BCOR ITD have been reported in 90% of cases of clear cell sarcoma of the kidney, with a smaller subset harboring YWHAE::NUTM2B/E or BCOR::CCNB3 gene fusions.[10,11] For more information, see the Clear Cell Sarcoma of the Kidney section in Wilms Tumor and Other Childhood Kidney Tumors Treatment.
The transcriptional profiles induced by BCOR gene fusions, BCOR ITD, and YWHAE::NUTM2B/E fusions appear to be similar to each other and distinctive from that of Ewing sarcoma.[2,6,7] As an example, elevated BCOR expression is observed across all of these entities, which can be useful in distinguishing these entities from other undifferentiated small round cell tumors.
Treatment of undifferentiated round cell sarcomas withBCORgenetic alterations
When treated with Ewing sarcoma–like therapies, 75% of patients show significant treatment-associated pathological responses. In one series of 36 cases, the 3-year and 5-year survival rates were 93% and 72%, respectively.[2][Level of evidence C1] In another series of 26 patients, the 5-year overall survival (OS) rate was 76.5%, and survival was better for patients who received induction therapy using an Ewing sarcoma–type regimen.[12][Level of evidence C1] Most of the tumors in these series arose in the bone. A retrospective survey of European cancer centers identified 148 patients with undifferentiated small round cell sarcomas who did not have an Ewing sarcoma–related fusion gene.[13] Of the 148 patients, 88 (60%) had CIC-rearranged sarcomas (median age, 32 years; range 7–78 years), 33 (22%) had BCOR::CCNB3-rearranged sarcomas (median age, 17 years; range 5–91 years), and 27 (18%) had unclassified undifferentiated small round cell sarcomas (median age, 37 years; range 4–70 years). Of the 148 patients, 101 (68.2%) presented with localized disease and 47 (31.8%) had metastasis at diagnosis. Male prevalence, younger age, bone primary site, and low rate of synchronous metastases were observed in BCOR::CCNB3-rearranged cases. The local treatment was surgery for 67 patients (45%) and surgery and radiation therapy for 52 patients (35%). Chemotherapy was given to 122 patients (82%). At a median follow-up of 42.7 months, the 3-year OS rate was 92.2% for patients with BCOR::CCNB3-rearranged sarcomas, 39.6% for patients with CIC-rearranged sarcomas, and 78.7% (P < .0001) for patients with unclassified undifferentiated small round cell sarcomas.
A multi-institution retrospective analysis of patients aged 0 to 24 years identified 29 patients with sarcomas and CIC gene fusions and 25 patients with BCOR-associated sarcomas (18 with BCOR::CCNB3 gene fusions and 7 with BCOR ITD).[14] Using a diverse range of treatments, the 3-year event-free survival (EFS) rates were 44.0% (95% confidence interval [CI], 28.7%–67.5%) for patients with CIC gene fusions and 41.2% (95% CI, 25.4%–67.0%) for patients with BCOR alterations (P = .97).
Undifferentiated Small Round Cell Sarcomas WithCICGenetic Alterations
CIC-rearranged sarcomas represent the second most common family of round cell sarcomas and are defined by the presence of CIC fusions at the molecular level.[1]
Clinical presentation
Undifferentiated small round cell sarcomas with CIC::DUX4 rearrangements most commonly affect young adults, with 50% of cases occurring between the ages of 21 and 40 years. In a series of 115 cases, the median age at diagnosis was 32 years, and 22% of cases occurred in patients younger than 18 years.[3,15] This entity more commonly affects males and usually originates from the soft tissues of the trunk and extremities.
Genomic characteristics
CIC-rearranged sarcomas most commonly have a CIC gene fusion with DUX4, FOXO4, or NUTM1.[15,16,17] The CIC gene fusion with DUX4 results from either a t(4;19)(q35;q13) or a t(10;19)(q26;q13) translocation.[16,18]CIC is located at chromosome 19q13.1 and DUX4 is located on either chromosome 4q35 or 10q26.3. Sarcomas with the CIC::DUX4 rearrangement have a transcriptional profile and DNA methylation profile that differs from that of Ewing sarcoma, supporting their characterization as a distinct entity.[6,19,20] For example, nearly all sarcomas with CIC::DUX4 rearrangements express WT1 and ETV4, in contrast to Ewing sarcoma and BCOR-rearranged tumors, making immunohistochemistry for these proteins useful in distinguishing between these diagnoses.[15,19]
Treatment of undifferentiated small round cell sarcomas withCICgenetic alterations
In a series of 115 cases of CIC-rearranged small round cell sarcomas, 57 patients had adequate follow-up information.[15] Nine patients presented with metastases, and 53% of patients with localized disease experienced a recurrence commonly involving the lung. Patients treated with neoadjuvant chemotherapy had an inferior survival than patients who were treated with up-front surgical resection. However, this difference in survival might have been related to a larger tumor size at presentation in the former group. The 2-year and 5-year survival rates were 53% and 43%, respectively.
An international retrospective cohort study further highlighted the poor outcomes for patients with CIC-rearranged sarcomas. The 3-year OS rate was 39.6%, which was significantly worse than outcomes for patients with other undifferentiated round cell sarcomas.[13] Likewise, these survival rates are significantly lower than the survival rates observed in patients with Ewing sarcoma. Further study is required to identify optimal treatments for this disease.
In another series of 79 patients with CIC-rearranged round cell sarcomas, outcomes were likewise poor, with a median OS of 18 months.[21] Patients treated with Ewing sarcoma–based chemotherapy regimens had nominally higher response rates compared with patients treated with soft tissue sarcoma–based regimens. However, OS rates were similar between these two groups.
A multi-institution retrospective analysis of patients aged 0 to 24 years identified 29 patients with sarcomas and CIC gene fusions and 25 patients with BCOR-associated sarcomas (18 with BCOR::CCNB3 gene fusions and 7 with BCOR ITD).[14] Using a diverse range of treatments, the 3-year EFS rates were 44.0% (95% CI, 28.7%–67.5%) for patients with CIC gene fusions and 41.2% (95% CI, 25.4%–67.0%) for patients with BCOR alterations (P = .97).
Undifferentiated small round cell sarcomas withCIC::NUTM1rearrangements
Undifferentiated small round cell sarcomas with CIC::NUTM1 rearrangements occur much less frequently than undifferentiated round cell sarcomas with CIC::DUX4 rearrangements.[22,23,24,25] These tumors occur in younger patients, with a median age of 6 years, compared with an average age of 21.6 years for patients with CIC::DUX4 fusions. The primary tumors occur in the central nervous system (CNS) and in the periphery. The histological appearance of these tumors is similar to CIC::DUX4-rearranged sarcomas. The prognosis of patients with these tumors is reported to be very poor despite treatment with surgery, multiagent chemotherapy, and radiation therapy.
Undifferentiated small round cell sarcomas withATXN1::NUTM2AorATXN1L::NUTM2Afusions
In one report, three children had tumors with ATXN1::NUTM2A or ATXN1L::NUTM2A fusions.[26] Two of the patients were infants with CNS lesions, and the third patient was a neonate with skin involvement and multiple masses throughout the peritoneal cavity. The authors suggested that ATXN1- or ATXN1L-associated fusions disrupted their interaction with CIC and decreased the transcription repressor complex, leading to downstream PEA3 family gene overexpression.
Undifferentiated Small Round Cell Sarcomas WithEWSR1::non-ETS Fusions
Sarcomas withEWSR1::NFATC2andFUS::NFATC2fusions
Sarcomas with EWSR1::NFATC2 and FUS::NFATC2 fusions typically arise in long bones, show a strong male predominance, and are more common in adults than in children.[27,28] These entities have transcriptional and DNA methylation profiles that distinguish them from Ewing sarcoma and other small round cell sarcomas.[6,20] Additionally, the transcriptional profiles for EWSR1::NFATC2 and FUS::NFATC2 differ from each other,[6] although the significance of this observation is unclear. The two entities also differ in that amplification of the EWSR1::NFATC2 gene fusion is commonly observed, but the FUS::NFATC2 gene fusion is generally not amplified.[20,27,29] Sarcomas with EWSR1::NFATC2 and FUS::NFATC2 fusions have metastatic potential and appear to respond poorly to chemotherapy regimens that are commonly used to treat other sarcomas.[27,28]
EWSR1::NFATC2 and FUS::NFATC2 rearrangements are also observed in a substantial proportion of solitary bone cysts (also known as simple bone cysts), a benign condition that typically presents in the metadiaphyses of the long bones of skeletally immature individuals.[30,31] Therefore, the presence of either EWSR1::NFATC2 or FUS::NFATC2 fusions should not be taken as an indicator of malignancy, but rather needs to be interpreted considering the clinical setting.
Sarcomas withEWSR1::PATZ1fusions
Sarcomas with the EWSR1::PATZ1 fusion are very uncommon. In the small number of cases described, there appears to be gender balance, a propensity for presentation at truncal primary sites (particularly the chest), and a median age of presentation of between 40 to 50 years, with cases rarely occurring in the pediatric age range.[32,33,34] Sarcomas with the EWSR1::PATZ1 fusion have gene expression and DNA methylation profiles that distinguish them from other sarcomas,[6,20] and CDKN2A deletions appear to commonly occur as secondary genomic alterations.[32,33]
The EWSR1::PATZ1 fusion has been described more commonly in brain tumors. It has been suggested that this fusion may define a novel form of glioblastoma.[35] In a series of 11 cases of EWSR1::PATZ1 fusion–associated tumors, 3 were primary brain tumors, 7 were sarcomas, and 1 was classified as a soft tissue sarcoma in the CNS.[32] Patients were between the ages of 11 and 81 years. Treatment details were reported for only three adult patients, two of whom had mixed responses to chemotherapy followed by disease progression, and one patient who did not receive chemotherapy.
References:
- WHO Classification of Tumours Editorial Board: WHO Classification of Tumours. Volume 3: Soft Tissue and Bone Tumours. 5th ed., IARC Press, 2020.
- Kao YC, Owosho AA, Sung YS, et al.: BCOR-CCNB3 Fusion Positive Sarcomas: A Clinicopathologic and Molecular Analysis of 36 Cases With Comparison to Morphologic Spectrum and Clinical Behavior of Other Round Cell Sarcomas. Am J Surg Pathol 42 (5): 604-615, 2018.
- Machado I, Navarro S, Llombart-Bosch A: Ewing sarcoma and the new emerging Ewing-like sarcomas: (CIC and BCOR-rearranged-sarcomas). A systematic review. Histol Histopathol 31 (11): 1169-81, 2016.
- Pierron G, Tirode F, Lucchesi C, et al.: A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat Genet 44 (4): 461-6, 2012.
- Specht K, Zhang L, Sung YS, et al.: Novel BCOR-MAML3 and ZC3H7B-BCOR Gene Fusions in Undifferentiated Small Blue Round Cell Sarcomas. Am J Surg Pathol 40 (4): 433-42, 2016.
- Watson S, Perrin V, Guillemot D, et al.: Transcriptomic definition of molecular subgroups of small round cell sarcomas. J Pathol 245 (1): 29-40, 2018.
- Kao YC, Sung YS, Zhang L, et al.: Recurrent BCOR Internal Tandem Duplication and YWHAE-NUTM2B Fusions in Soft Tissue Undifferentiated Round Cell Sarcoma of Infancy: Overlapping Genetic Features With Clear Cell Sarcoma of Kidney. Am J Surg Pathol 40 (8): 1009-20, 2016.
- Kao YC, Sung YS, Zhang L, et al.: BCOR Overexpression Is a Highly Sensitive Marker in Round Cell Sarcomas With BCOR Genetic Abnormalities. Am J Surg Pathol 40 (12): 1670-1678, 2016.
- Antonescu CR, Kao YC, Xu B, et al.: Undifferentiated round cell sarcoma with BCOR internal tandem duplications (ITD) or YWHAE fusions: a clinicopathologic and molecular study. Mod Pathol 33 (9): 1669-1677, 2020.
- Ueno-Yokohata H, Okita H, Nakasato K, et al.: Consistent in-frame internal tandem duplications of BCOR characterize clear cell sarcoma of the kidney. Nat Genet 47 (8): 861-3, 2015.
- Roy A, Kumar V, Zorman B, et al.: Recurrent internal tandem duplications of BCOR in clear cell sarcoma of the kidney. Nat Commun 6: 8891, 2015.
- Cohen-Gogo S, Cellier C, Coindre JM, et al.: Ewing-like sarcomas with BCOR-CCNB3 fusion transcript: a clinical, radiological and pathological retrospective study from the Société Française des Cancers de L'Enfant. Pediatr Blood Cancer 61 (12): 2191-8, 2014.
- Palmerini E, Gambarotti M, Italiano A, et al.: A global collaboRAtive study of CIC-rearranged, BCOR::CCNB3-rearranged and other ultra-rare unclassified undifferentiated small round cell sarcomas (GRACefUl). Eur J Cancer 183: 11-23, 2023.
- Sparber-Sauer M, Corradini N, Affinita MC, et al.: Clinical characteristics and outcomes for children, adolescents and young adults with "CIC-fused" or "BCOR-rearranged" soft tissue sarcomas: A multi-institutional European retrospective analysis. Cancer Med 12 (13): 14346-14359, 2023.
- Antonescu CR, Owosho AA, Zhang L, et al.: Sarcomas With CIC-rearrangements Are a Distinct Pathologic Entity With Aggressive Outcome: A Clinicopathologic and Molecular Study of 115 Cases. Am J Surg Pathol 41 (7): 941-949, 2017.
- Italiano A, Sung YS, Zhang L, et al.: High prevalence of CIC fusion with double-homeobox (DUX4) transcription factors in EWSR1-negative undifferentiated small blue round cell sarcomas. Genes Chromosomes Cancer 51 (3): 207-18, 2012.
- Dickson BC, Sung YS, Rosenblum MK, et al.: NUTM1 Gene Fusions Characterize a Subset of Undifferentiated Soft Tissue and Visceral Tumors. Am J Surg Pathol 42 (5): 636-645, 2018.
- Kawamura-Saito M, Yamazaki Y, Kaneko K, et al.: Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum Mol Genet 15 (13): 2125-37, 2006.
- Specht K, Sung YS, Zhang L, et al.: Distinct transcriptional signature and immunoprofile of CIC-DUX4 fusion-positive round cell tumors compared to EWSR1-rearranged Ewing sarcomas: further evidence toward distinct pathologic entities. Genes Chromosomes Cancer 53 (7): 622-33, 2014.
- Koelsche C, Kriegsmann M, Kommoss FKF, et al.: DNA methylation profiling distinguishes Ewing-like sarcoma with EWSR1-NFATc2 fusion from Ewing sarcoma. J Cancer Res Clin Oncol 145 (5): 1273-1281, 2019.
- Brahmi M, Gaspar N, Gantzer J, et al.: Patterns of care and outcome of CIC-rearranged sarcoma patients: A nationwide study of the French sarcoma group. Cancer Med 12 (7): 7801-7807, 2023.
- Le Loarer F, Pissaloux D, Watson S, et al.: Clinicopathologic Features of CIC-NUTM1 Sarcomas, a New Molecular Variant of the Family of CIC-Fused Sarcomas. Am J Surg Pathol 43 (2): 268-276, 2019.
- Mangray S, Kelly DR, LeGuellec S, et al.: Clinicopathologic Features of a Series of Primary Renal CIC-rearranged Sarcomas With Comprehensive Molecular Analysis. Am J Surg Pathol 42 (10): 1360-1369, 2018.
- Schaefer IM, Dal Cin P, Landry LM, et al.: CIC-NUTM1 fusion: A case which expands the spectrum of NUT-rearranged epithelioid malignancies. Genes Chromosomes Cancer 57 (9): 446-451, 2018.
- Zhao L, He H, Ren J, et al.: CIC::NUTM1 sarcomas occurred in soft tissues of upper limbs : a rare case report and literature review. Diagn Pathol 19 (1): 76, 2024.
- Xu F, Viaene AN, Ruiz J, et al.: Novel ATXN1/ATXN1L::NUTM2A fusions identified in aggressive infant sarcomas with gene expression and methylation patterns similar to CIC-rearranged sarcoma. Acta Neuropathol Commun 10 (1): 102, 2022.
- Bode-Lesniewska B, Fritz C, Exner GU, et al.: EWSR1-NFATC2 and FUS-NFATC2 Gene Fusion-Associated Mesenchymal Tumors: Clinicopathologic Correlation and Literature Review. Sarcoma 2019: 9386390, 2019.
- Wang GY, Thomas DG, Davis JL, et al.: EWSR1-NFATC2 Translocation-associated Sarcoma Clinicopathologic Findings in a Rare Aggressive Primary Bone or Soft Tissue Tumor. Am J Surg Pathol 43 (8): 1112-1122, 2019.
- Szuhai K, Ijszenga M, de Jong D, et al.: The NFATc2 gene is involved in a novel cloned translocation in a Ewing sarcoma variant that couples its function in immunology to oncology. Clin Cancer Res 15 (7): 2259-68, 2009.
- Pižem J, Šekoranja D, Zupan A, et al.: FUS-NFATC2 or EWSR1-NFATC2 Fusions Are Present in a Large Proportion of Simple Bone Cysts. Am J Surg Pathol 44 (12): 1623-1634, 2020.
- Hung YP, Fisch AS, Diaz-Perez JA, et al.: Identification of EWSR1-NFATC2 fusion in simple bone cysts. Histopathology 78 (6): 849-856, 2021.
- Bridge JA, Sumegi J, Druta M, et al.: Clinical, pathological, and genomic features of EWSR1-PATZ1 fusion sarcoma. Mod Pathol 32 (11): 1593-1604, 2019.
- Michal M, Rubin BP, Agaimy A, et al.: EWSR1-PATZ1-rearranged sarcoma: a report of nine cases of spindle and round cell neoplasms with predilection for thoracoabdominal soft tissues and frequent expression of neural and skeletal muscle markers. Mod Pathol 34 (4): 770-785, 2021.
- Dehner CA, Torres-Mora J, Gupta S, et al.: Sarcomas Harboring EWSR1::PATZ1 Fusions: A Clinicopathologic Study of 17 Cases. Mod Pathol 37 (2): 100400, 2024.
- Siegfried A, Rousseau A, Maurage CA, et al.: EWSR1-PATZ1 gene fusion may define a new glioneuronal tumor entity. Brain Pathol 29 (1): 53-62, 2019.
Latest Updates to This Summary (11 / 27 / 2024)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Treatment of Recurrent Ewing Sarcoma
Added strategies to target fusion proteins to the list of other therapies that have been studied in the treatment of recurrent Ewing sarcoma.
Added text to state that lurbinectedin is structurally related to trabectedin, but it has been more effective in suppressing the activity of the oncogenic transcription factor EWS::FLI in mice in preclinical studies. Also added text about the results of an open-label, single-arm, basket phase II trial of lurbinectedin (cited Subbiah et al. as reference 68). Added text to state that an agent known as TK216 is thought to interfere with interactions between the EWSR1::FLI1 fusion oncoprotein and key proteins critical for its oncogenic function, although it has also been shown to disrupt microtubules (cited Povedano et al. as reference 69). Also added text about the results of a phase I/II trial of TK216 in combination with vincristine in patients with recurrent Ewing sarcoma (cited Meyers et al. as reference 70).
Added NCT04890093 as a treatment option under clinical evaluation for patients with recurrent Ewing sarcoma.
Undifferentiated Small Round Cell (Ewing-Like) Sarcomas
Added text to state that CIC-rearranged sarcomas represent the second most common family of round cell sarcomas and are defined by the presence of CIC fusions at the molecular level.
Revised text to state that CIC-rearranged sarcomas most commonly have a CIC gene fusion with DUX4, FOXO4, or NUTM1 (cited Dickson et al. as reference 17).
Added Zhao et al. as reference 25. Also revised text to state that tumors with CIC::NUTM1 rearrangements occur in younger patients, with a median age of 6 years, compared with an average age of 21.6 years for patients with CIC::DUX4 fusions.
Added Dehner et al. as reference 34.
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of childhood Ewing sarcoma and undifferentiated small round cell sarcomas of bone and soft tissue. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Ewing Sarcoma and Undifferentiated Small Round Cell Sarcomas of Bone and Soft Tissue Treatment are:
- Holcombe Edwin Grier, MD
- Andrea A. Hayes-Dixon, MD, FACS, FAAP (Howard University)
- Karen J. Marcus, MD, FACR (Dana-Farber of Boston Children's Cancer Center and Blood Disorders Harvard Medical School)
- William H. Meyer, MD
- Paul A. Meyers, MD (Memorial Sloan-Kettering Cancer Center)
- Thomas A. Olson, MD (Aflac Cancer and Blood Disorders Center of Children's Healthcare of Atlanta - Egleston Campus)
- Nita Louise Seibel, MD (National Cancer Institute)
- Malcolm A. Smith, MD, PhD (National Cancer Institute)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as "NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary]."
The preferred citation for this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Ewing Sarcoma and Undifferentiated Small Round Cell Sarcomas of Bone and Soft Tissue Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/bone/hp/ewing-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389480]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either "standard" or "under clinical evaluation." These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website's Email Us.
Last Revised: 2024-11-27

